| Фосфор(P) | |
|---|---|
| Атомный номер | 15 |
| Внешний вид простого вещества | Белый фосфор- белый, восковидный, слегка фосфоресцирующий |
| Свойства атома | |
| Атомная масса (молярная масса) |
30,973762 а. е. м. (г/моль) |
| Радиус атома | 128 пм |
| Энергия ионизации (первый электрон) |
1011,2(10,48) кДж/моль (эВ) |
| Электронная конфигурация | [Ne] 3s2 3p3 |
| Химические свойства | |
| Ковалентный радиус | 106 пм |
| Радиус иона | 35 (+5e) 212 (-3e) пм |
| Электроотрицательность (по Полингу) |
2,19 |
| Электродный потенциал | 0 |
| Степени окисления | 5, 3, -3 |
| Термодинамические свойства простого вещества | |
| Плотность | (белый фосфор)1,82 г/см³ |
| Удельная теплоёмкость | 0,757 Дж/(K·моль) |
| Теплопроводность | (0,236) Вт/(м·K) |
| Температура плавления | 317,3 K |
| Теплота плавления | 2,51 кДж/моль |
| Температура кипения | 553 K |
| Теплота испарения | 49,8 кДж/моль |
| Молярный объём | 17,0 см³/моль |
| Кристаллическая решётка простого вещества | |
| Структура решётки | кубическая |
| Период решётки | 7,170 Å |
| Отношение c/a | n/a |
| Температура Дебая | n/a K |
| P | 15 |
| 30,973762 | |
| 3s²3p³ | |
| Фосфор |
Фосфор — один из самых распространённых элементов земной коры, его содержание составляет 0,08—0,09 % её массы. В свободном состоянии не встречается из-за высокой химической активности. Образует около 190 минералов, важнейшими из которых являются апатит Ca5(PO4)3F, фосфорит Ca3(PO4)2 и другие. Фосфор содержится во всех частях зелёных растений, ещё больше его в плодах и семенах (см. фосфолипиды). Содержится в животных тканях, входит в состав белков и других важнейших органических соединений (АТФ), является элементом жизни.
Содержание
- 1 История
- 2 Происхождение названия
- 3 Получение
- 4 Физические свойства
- 4.1 Белый фосфор
- 4.1.1 Жёлтый фосфор
- 4.2 Красный фосфор
- 4.3 Чёрный фосфор
- 4.4 Металлический фосфор
- 4.1 Белый фосфор
- 5 Химические свойства
- 5.1 Взаимодействие с простыми веществами
- 5.2 Взаимодействие с водой
- 5.3 Взаимодействие со щелочами
- 5.4 Восстановительные свойства
- 6 Применение
- 6.1 Элементарный фосфор
- 6.2 Соединения фосфора в сельском хозяйстве
- 6.3 Соединения фосфора в промышленности
- 6.4 Фосфатные связующие
- 7 Биологическая роль соединений фосфора
- 7.1 Токсикология элементарного фосфора
- 7.2 Токсикология соединений фосфора
- 8 Примечания
- 9 См. также
- 10 Ссылки
История

Схема атома фосфора
Фосфор открыт гамбургским алхимиком Хеннингом Брандом в 1669 году. Подобно другим алхимикам, Бранд пытался отыскать эликсир жизни или философский камень, а получил светящееся вещество. Существуют данные, что фосфор умели получать еще арабские алхимики в XII в. То, что фосфор — простое вещество, доказал Лавуазье.
Происхождение названия
В 1669 Хеннинг Бранд при нагревании смеси белого песка и выпаренной мочи получил светящееся в темноте вещество, названное сначала «холодным огнём». Вторичное название «фосфор» происходит от греческих слов «фос» — свет и «феро» — несу.
Получение
Фосфор получают из апатитов или фосфоритов в результате взаимодействия с коксом и песком при температуре 1500 °С:
- 2Ca3(PO4)2 + 10C + 6SiO2 → 4P + 10CO + 6CaSiO3.
Образующиеся пары белого фосфора конденсируются в приёмнике под водой. Вместо фосфоритов восстановлению можно подвергнуть и другие соединения, например, метафосфорную кислоту:
- 4HPO3 + 12C → 4P + 2H2 + 12CO.
Физические свойства
Элементарный фосфор в обычных условиях представляет собой несколько устойчивых аллотропических модификаций; вопрос аллотропии фосфора сложен и до конца не решён. Обычно выделяют четыре модификации простого вещества — белую, красную, черную и металлический фосфор. Иногда их ещё называют главными аллотропными модификациями, подразумевая при этом, что все остальные являются разновидностью указанных четырёх. В обычных условиях существует только три аллотропических модификации фосфора, а в условиях сверхвысоких давлений — также металлическая форма. Все модификации различаются по цвету, плотности и другим физическим характеристикам; заметна тенденция к резкому убыванию химической активности при переходе от белого к металлическому фосфору и нарастанию металлических свойств.
Белый фосфор
Белый фосфор представляет собой белое вещество (из-за примесей может иметь желтоватый оттенок) с температурой плавления 44,1 °С. По внешнему виду он очень похож на очищенный воск или парафин, легко режется ножом и деформируется от небольших усилий. Отливаемый в инертной атмосфере в виде палочек (слитков), он сохраняется в отсутствии воздуха под слоем очищенной воды или в специальных инертных средах. Химически белый фосфор чрезвычайно активен. Например, белый фосфор медленно окисляется кислородом воздуха уже при комнатной температуре и светится (бледно-зелёное свечение). Явление такого рода свечения вследствие химических реакций окисления называется хемилюминесценцией или устаревшим термином — фосфоресценцией. Белый фосфор не только активен химически, но и весьма ядовит (вызывает поражение костей, костного мозга, некроз челюстей) и легкорастворим в органических растворителях. Летальная доза белого фосфора для взрослого мужчины составляет 0,05—0,1 г. Растворимостью белого фосфора в сероуглероде пользуются для промышленной очистки его от примесей. Плотность белого фосфора из всех его модификаций наименьшая и составляет около 1823 кг/м³.
Жёлтый фосфор
Неочищенный белый фосфор обычно называют «жёлтый фосфор». Сильноядовитое (ПДК 0,03 мг/м³), огнеопасное кристаллическое вещество от светло-жёлтого до тёмно-бурого цвета. Удельный вес 1,83 г/см³, плавится при +34 °C, кипит при +280 °C. В воде не растворяется, на воздухе легко окисляется и самовоспламеняется. Горит с выделением густого белого дыма — мелких частичек декаоксида тетрафосфора P4O10[1]. Несмотря на то, что в результате реакции между фосфором и водой (4Р + 6Н2О → РН3 + 3Н3РО2) выделяется ядовитый газ фосфин (РН3), для тушения фосфора используют воду в больших количествах (для снижения температуры очага возгорания и перевода фосфора в твердое состояние) или раствор сульфата меди (медного купороса), после гашения фосфор засыпают влажным песком. Для предохранения от самовозгорания желтый фосфор хранится и перевозится под слоем воды (раствора хлорида кальция).
Красный фосфор

Красный фосфор, также называемый фиолетовым фосфором, — это более термодинамически стабильная модификация элементарного фосфора. Впервые он был получен в 1847 году в Швеции австрийским химиком А. Шрёттером при нагревании белого фосфора при 500 °С в атмосфере угарного газа (СО) в запаянной стеклянной ампуле.
Красный фосфор имеет формулу (Р4)n и представляет собой полимер со сложной структурой. В зависимости от способа получения и степени дробления красного фосфора, имеет оттенки от пурпурно-красного до фиолетового, а в литом состоянии — тёмно-фиолетовый с медным оттенком металлический блеск. Химическая активность красного фосфора значительно ниже, чем у белого; ему присуща исключительно малая растворимость. Растворить красный фосфор возможно лишь в некоторых расплавленных металлах (свинец и висмут), чем иногда пользуются для получения крупных его кристаллов. Так, например, немецкий физико-химик И. В. Гитторф в 1865 году впервые получил прекрасно построенные, но небольшие по размеру кристаллы (фосфор Гитторфа). На воздухе красный фосфор воспламеняется при высоких температурах (при переходе в белую форму во время возгонки), и у него полностью отсутствует явление хемолюминесценции. Ядовитость его в тысячи раз меньше, чем у белого, поэтому он применяется гораздо шире, например, в производстве спичек. Плотность красного фосфора также выше, и достигает 2400 кг/м³ в литом виде. При хранении на воздухе красный фосфор в присутствии влаги постепенно окисляется, образуя гигроскопичный оксид, поглощает воду и отсыревает («отмокает»), образуя вязкую фосфорную кислоту; поэтому его хранят в герметичной таре. При «отмокании» — промывают водой от остатков фосфорных кислот, высушивают и используют по назначению.
Чёрный фосфор
Чёрный фосфор — это наиболее стабильная термодинамически и химически наименее активная форма элементарного фосфора. Впервые чёрный фосфор был получен в 1914 году американским физиком П. У. Бриджменом из белого фосфора в виде чёрных блестящих кристаллов, имеющих высокую (2690 кг/м³) плотность. Для проведения синтеза чёрного фосфора Бриджмен применил давление в 2·109 Па (20 тысяч атмосфер) и температуру около 200 °С. Начало быстрого перехода лежит в области 13 000 атмосфер и температуре около 230 °С.
Чёрный фосфор представляет собой чёрное вещество с металлическим блеском, жирное на ощупь и весьма похожее на графит, и с полностью отсутствующей растворимостью в воде или органических растворителях. Поджечь чёрный фосфор можно, только предварительно сильно раскалив в атмосфере чистого кислорода до 400 °С. Удивительным свойством чёрного фосфора является его способность проводить электрический ток и свойства полупроводника. Температура плавления чёрного фосфора 1000 °С под давлением 18·105 Па.
Металлический фосфор
При 8,3·1010 Па чёрный фосфор переходит в новую, ещё более плотную и инертную металлическую фазу с плотностью 3,56 г/см³, а при дальнейшем повышении давления до 1,25·1011 Па — ещё более уплотняется и приобретает кубическую кристаллическую решётку, при этом его плотность возрастает до 3,83 г/см³. Металлический фосфор очень хорошо проводит электрический ток.
Химические свойства
Химическая активность фосфора значительно выше, чем у азота. Химические свойства фосфора во многом определяются его аллотропной модификацией. Белый фосфор очень активен, в процессе перехода к красному и чёрному фосфору химическая активность резко снижается. Белый фосфор на воздухе светится в темноте, свечение обусловлено окислением паров фосфора до низших оксидов.
В жидком и растворенном состоянии, а также в парах до 800 °С фосфор состоит из молекул Р4. При нагревании выше 800 °С молекулы диссоциируют: Р4 = 2Р2. При температуре выше 2000 °С молекулы распадаются на атомы.
Взаимодействие с простыми веществами
Фосфор легко окисляется кислородом:
- 4P + 5O2 → 2P2O5,
- 4P + 3O2 → 2P2O3.
Взаимодействует со многими простыми веществами — галогенами, серой, некоторыми металлами, проявляя окислительные и восстановительные свойства:
с металлами — окислитель, образует фосфиды:
- 2P + 3Ca → Ca3P2.
- 2P + 3Mg → Mg3P2.
с неметаллами — восстановитель:
- 2P + 3S → P2S3,
- 2P + 3Cl2 → 2PCl3.
Не взаимодействует с водородом.
Взаимодействие с водой
Взаимодействует с водой, при этом диспропорционирует:
- 4Р + 6Н2О → РН3 + 3Н3РО2 (фосфатная кислота).
Взаимодействие со щелочами
В растворах щелочей диспропорционирование происходит в большей степени:
- 4Р + 3KOH + 3Н2О → РН3 + 3KН2РО2.
Восстановительные свойства
Сильные окислители превращают фосфор в фосфорную кислоту:
- 3P + 5HNO3 + 2H2O → 3H3PO4 + 5NO;
- 2P + 5H2SO4 → 2H3PO4 + 5SO2 + 2H2O.
Реакция окисления также происходит при поджигании спичек, в качестве окислителя выступает бертолетова соль:
- 6P + 5KClO3 → 5KCl + 3P2O5
Применение
Фосфор является важнейшим биогенным элементом и в то же время находит очень широкое применение в промышленности. Красный фосфор применяют в производстве спичек. Его вместе с тонко измельчённым стеклом и клеем наносят на боковую поверхность коробка. При трении спичечной головки в состав который входят хлорат калия и сера, происходит воспламенение.
Элементарный фосфор
Пожалуй, первое свойство фосфора, которое человек поставил себе на службу, — это горючесть. Горючесть фосфора очень велика и зависит от аллотропической модификации.
Наиболее активен химически, токсичен и горюч белый («жёлтый») фосфор, потому он очень часто применяется (в зажигательных бомбах и пр.).
Красный фосфор — основная модификация, производимая и потребляемая промышленностью. Он применяется в производстве спичек, взрывчатых веществ, зажигательных составов, топлив, а также противозадирных смазочных материалов.
Соединения фосфора в сельском хозяйстве
Фосфор (в виде фосфатов) — один из трёх важнейших биогенных элементов (NPK), участвует в синтезе АТФ. Большая часть производимой фосфорной кислоты идёт на получение фосфорных удобрений — суперфосфата, преципитата, аммофоски и др.
Соединения фосфора в промышленности
Фосфаты широко используются:
- в качестве комплексообразователей (средства для умягчения воды),
- в составе пассиваторов поверхности металлов (защита от коррозии, например, т. н. состав «мажеф»),
Фосфатные связующие
Способность фосфатов формировать прочную трёхмерную полимерную сетку используется для изготовления фосфатных и алюмофосфатных связок
Биологическая роль соединений фосфора
Фосфор присутствует в живых клетках в виде орто- и пирофосфорной кислот, входит в состав нуклеотидов, нуклеиновых кислот, фосфопротеидов, фосфолипидов, коферментов, ферментов. Кости человека состоят из гидроксилапатита 3Са3(РО4)3·CaF2. В состав зубной эмали входит фторапатит. Основную роль в превращениях соединений фосфора в организме человека и животных играет печень. Обмен фосфорных соединений регулируется гормонами и витамином D. Суточная потребность человека в фосфоре 800-1500 мг. При недостатке фосфора в организме развиваются различные заболевания костей.
Токсикология элементарного фосфора
- Красный фосфор практически нетоксичен. Пыль красного фосфора, попадая в легкие, вызывает пневмонию при хроническом действии.
- Белый фосфор очень ядовит, растворим в липидах. Смертельная доза белого фосфора — 50-150 мг. Попадая на кожу, белый фосфор дает тяжелые ожоги.
Острые отравления фосфором проявляются жжением во рту и желудке, головной болью, слабостью, рвотой. Через 2-3 суток развивается желтуха. Для хронических форм характерны нарушение кальциевого обмена, поражение сердечно-сосудистой и нервной систем. Первая помощь при остром отравлении — промывание желудка, слабительное, очистительные клизмы, внутривенно растворы глюкозы. При ожогах кожи обработать пораженные участки растворами медного купороса или соды. ПДК паров фосфора в воздухе 0,03 мг/м3.
Токсикология соединений фосфора
Некоторые соединения фосфора (фосфин) очень токсичны. Боевые отравляющие вещества зарин, зоман, табун являются соединениями фосфора.
Примечания
- ↑ В. Шретер, К.-Х. Лаутеншлегер, Х. Бибрак и др. Химия = Chemie. — М.: Химия, 1989. — С. 351. — ISBN 5-7245-0360-3
См. также
- Категория:Соединения фосфора
Ссылки
- Фосфор на Webelements
- Фосфор в Популярной библиотеке химических элементов
- Российские ученые лишились радиоактивной метки, polit.ru, 5 мая 2008
Wikimedia Foundation.
2010.
|
Waxy white
Light red
Dark red and violet |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Phosphorus | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pronunciation | (FOS-fər-əs) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Allotropes | white, red, violet, black and others (see Allotropes of phosphorus) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Appearance | white, red and violet are waxy, black is metallic-looking | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Standard atomic weight Ar°(P) |
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Abundance | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| in the Earth’s crust | 5.2 (silicon = 100) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Phosphorus in the periodic table | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atomic number (Z) | 15 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Group | group 15 (pnictogens) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Period | period 3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Block | p-block | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electron configuration | [Ne] 3s2 3p3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electrons per shell | 2, 8, 5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Physical properties | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Phase at STP | solid | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Melting point | white: 317.3 K (44.15 °C, 111.5 °F) red: ∼860 K (∼590 °C, ∼1090 °F)[2] |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Boiling point | white: 553.7 K (280.5 °C, 536.9 °F) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Sublimation point | red: ≈689.2–863 K (≈416–590 °C, ≈780.8–1094 °F) violet: 893 K (620 °C, 1148 °F) |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density (near r.t.) | white: 1.823 g/cm3 red: ≈2.2–2.34 g/cm3 violet: 2.36 g/cm3 black: 2.69 g/cm3 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Heat of fusion | white: 0.66 kJ/mol | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Heat of vaporisation | white: 51.9 kJ/mol | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Molar heat capacity | white: 23.824 J/(mol·K) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Vapour pressure (white)
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Vapour pressure (red, b.p. 431 °C)
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atomic properties | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Oxidation states | −3, −2, −1, 0,[3] +1,[4] +2, +3, +4, +5 (a mildly acidic oxide) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electronegativity | Pauling scale: 2.19 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Ionisation energies |
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Covalent radius | 107±3 pm | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Van der Waals radius | 180 pm | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Spectral lines of phosphorus |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Other properties | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Natural occurrence | primordial | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal structure | body-centred cubic (bcc)
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Thermal conductivity | white: 0.236 W/(m⋅K) black: 12.1 W/(m⋅K) |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Magnetic ordering | white, red, violet, black: diamagnetic[5] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Molar magnetic susceptibility | −20.8×10−6 cm3/mol (293 K)[6] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Bulk modulus | white: 5 GPa red: 11 GPa |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CAS Number | 7723-14-0 (red) 12185-10-3 (white) |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| History | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Discovery | Hennig Brand (1669) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Recognised as an element by | Antoine Lavoisier[7] (1777) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Main isotopes of phosphorus
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| references |




Phosphorus is a chemical element with the symbol P and atomic number 15. Elemental phosphorus exists in two major forms, white phosphorus and red phosphorus, but because it is highly reactive, phosphorus is never found as a free element on Earth. It has a concentration in the Earth’s crust of about one gram per kilogram (compare copper at about 0.06 grams). In minerals, phosphorus generally occurs as phosphate.
Elemental phosphorus was first isolated as white phosphorus in 1669. White phosphorus emits a faint glow when exposed to oxygen – hence the name, taken from Greek mythology, Φωσφόρος meaning ‘light-bearer’ (Latin Lucifer), referring to the «Morning Star», the planet Venus. The term phosphorescence, meaning glow after illumination, derives from this property of phosphorus, although the word has since been used for a different physical process that produces a glow. The glow of phosphorus is caused by oxidation of the white (but not red) phosphorus — a process now called chemiluminescence. Together with nitrogen, arsenic, antimony, and bismuth, phosphorus is classified as a pnictogen.
Phosphorus is an element essential to sustaining life largely through phosphates, compounds containing the phosphate ion, PO43−. Phosphates are a component of DNA, RNA, ATP, and phospholipids, complex compounds fundamental to cells. Elemental phosphorus was first isolated from human urine, and bone ash was an important early phosphate source. Phosphate mines contain fossils because phosphate is present in the fossilized deposits of animal remains and excreta. Low phosphate levels are an important limit to growth in some aquatic systems. The vast majority of phosphorus compounds mined are consumed as fertilisers. Phosphate is needed to replace the phosphorus that plants remove from the soil, and its annual demand is rising nearly twice as fast as the growth of the human population. Other applications include organophosphorus compounds in detergents, pesticides, and nerve agents.
Characteristics[edit]
Allotropes[edit]
Phosphorus has several allotropes that exhibit strikingly diverse properties.[8] The two most common allotropes are white phosphorus and red phosphorus.[9]
From the perspective of applications and chemical literature, the most important form of elemental phosphorus is white phosphorus, often abbreviated as WP. It is a soft, waxy solid which consists of tetrahedral P
4 molecules, in which each atom is bound to the other three atoms by a formal single bond. This P
4 tetrahedron is also present in liquid and gaseous phosphorus up to the temperature of 800 °C (1,470 °F) when it starts decomposing to P
2 molecules.[10] The P
4 molecule in the gas phase has a P-P bond length of rg = 2.1994(3) Å as was determined by gas electron diffraction.[11] The nature of bonding in this P
4 tetrahedron can be described by spherical aromaticity or cluster bonding, that is the electrons are highly delocalized. This has been illustrated by calculations of the magnetically induced currents, which sum up to 29 nA/T, much more than in the archetypical aromatic molecule benzene (11 nA/T).[11]
White phosphorus exists in two crystalline forms: α (alpha) and β (beta). At room temperature, the α-form is stable. It is more common, has cubic crystal structure and at 195.2 K (−78.0 °C), it transforms into β-form, which has hexagonal crystal structure. These forms differ in terms of the relative orientations of the constituent P4 tetrahedra.[12][13] The β form of white phosphorus contains three slightly different P
4 molecules, i.e. 18 different P-P bond lengths between 2.1768(5) and 2.1920(5) Å. The average P-P bond length is 2.183(5) Å.[14]
White phosphorus is the least stable, the most reactive, the most volatile, the least dense and the most toxic of the allotropes. White phosphorus gradually changes to red phosphorus. This transformation is accelerated by light and heat, and samples of white phosphorus almost always contain some red phosphorus and accordingly appear yellow. For this reason, white phosphorus that is aged or otherwise impure (e.g., weapons-grade, not lab-grade WP) is also called yellow phosphorus. When exposed to oxygen, white phosphorus glows in the dark with a very faint tinge of green and blue. It is highly flammable and pyrophoric (self-igniting) upon contact with air. Owing to its pyrophoricity, white phosphorus is used as an additive in napalm. The odour of combustion of this form has a characteristic garlic smell, and samples are commonly coated with white phosphorus pentoxide, which consists of P
4O
10 tetrahedra with oxygen inserted between the phosphorus atoms and at their vertices. White phosphorus is insoluble in water but soluble in carbon disulfide.[15]
Thermal decomposition of P4 at 1100 K gives diphosphorus, P2. This species is not stable as a solid or liquid. The dimeric unit contains a triple bond and is analogous to N2. It can also be generated as a transient intermediate in solution by thermolysis of organophosphorus precursor reagents.[16] At still higher temperatures, P2 dissociates into atomic P.[15]
| Form | white(α) | white(β) | red | violet | black |
|---|---|---|---|---|---|
| Symmetry | Body-centred cubic |
Triclinic | Amorphous | Monoclinic | Orthorhombic |
| Pearson symbol | aP24 | mP84 | oS8 | ||
| Space group | I43m | P1 No.2 | P2/c No.13 | Cmca No.64 | |
| Density (g/cm3) | 1.828 | 1.88 | ~2.2 | 2.36 | 2.69 |
| Band gap (eV) | 2.1 | 1.8 | 1.5 | 0.34 | |
| Refractive index | 1.8244 | 2.6 | 2.4 |
Red phosphorus is polymeric in structure. It can be viewed as a derivative of P4 wherein one P-P bond is broken, and one additional bond is formed with the neighbouring tetrahedron resulting in chains of P21 molecules linked by van der Waals forces.[18] Red phosphorus may be formed by heating white phosphorus to 250 °C (482 °F) or by exposing white phosphorus to sunlight.[19] Phosphorus after this treatment is amorphous. Upon further heating, this material crystallises. In this sense, red phosphorus is not an allotrope, but rather an intermediate phase between the white and violet phosphorus, and most of its properties have a range of values. For example, freshly prepared, bright red phosphorus is highly reactive and ignites at about 300 °C (572 °F),[20] though it is more stable than white phosphorus, which ignites at about 30 °C (86 °F).[21] After prolonged heating or storage, the color darkens (see infobox images); the resulting product is more stable and does not spontaneously ignite in air.[22]
Violet phosphorus is a form of phosphorus that can be produced by day-long annealing of red phosphorus above 550 °C. In 1865, Hittorf discovered that when phosphorus was recrystallised from molten lead, a red/purple form is obtained. Therefore, this form is sometimes known as «Hittorf’s phosphorus» (or violet or α-metallic phosphorus).[17]
Black phosphorus is the least reactive allotrope and the thermodynamically stable form below 550 °C (1,022 °F). It is also known as β-metallic phosphorus and has a structure somewhat resembling that of graphite.[23][24] It is obtained by heating white phosphorus under high pressures (about 12,000 standard atmospheres or 1.2 gigapascals). It can also be produced at ambient conditions using metal salts, e.g. mercury, as catalysts.[25] In appearance, properties, and structure, it resembles graphite, being black and flaky, a conductor of electricity, and has puckered sheets of linked atoms.[26]
Another form, scarlet phosphorus, is obtained by allowing a solution of white phosphorus in carbon disulfide to evaporate in sunlight.[17]
Chemiluminescence[edit]
White phosphorus exposed to air glows in the dark
When first isolated, it was observed that the green glow emanating from white phosphorus would persist for a time in a stoppered jar, but then cease. Robert Boyle in the 1680s ascribed it to «debilitation» of the air. Actually, it is oxygen being consumed. By the 18th century, it was known that in pure oxygen, phosphorus does not glow at all;[27] there is only a range of partial pressures at which it does. Heat can be applied to drive the reaction at higher pressures.[28]
In 1974, the glow was explained by R. J. van Zee and A. U. Khan.[29][30] A reaction with oxygen takes place at the surface of the solid (or liquid) phosphorus, forming the short-lived molecules HPO and P
2O
2 that both emit visible light. The reaction is slow and only very little of the intermediates are required to produce the luminescence, hence the extended time the glow continues in a stoppered jar.
Since its discovery, phosphors and phosphorescence were used loosely to describe substances that shine in the dark without burning. Although the term phosphorescence is derived from phosphorus, the reaction that gives phosphorus its glow is properly called chemiluminescence (glowing due to a cold chemical reaction), not phosphorescence (re-emitting light that previously fell onto a substance and excited it).[31]
Isotopes[edit]
There are 23 known isotopes of phosphorus,[32] ranging from 25
P to 47
P.[33] Only 31
P is stable and is therefore present at 100% abundance. The half-integer nuclear spin and high abundance of 31P make phosphorus-31 NMR spectroscopy a very useful analytical tool in studies of phosphorus-containing samples.
Two radioactive isotopes of phosphorus have half-lives suitable for biological scientific experiments. These are:
- 32
P, a beta-emitter (1.71 MeV) with a half-life of 14.3 days, which is used routinely in life-science laboratories, primarily to produce radiolabeled DNA and RNA probes, e.g. for use in Northern blots or Southern blots. - 33
P, a beta-emitter (0.25 MeV) with a half-life of 25.4 days. It is used in life-science laboratories in applications in which lower energy beta emissions are advantageous such as DNA sequencing.
The high energy beta particles from 32
P penetrate skin and corneas and any 32
P ingested, inhaled, or absorbed is readily incorporated into bone and nucleic acids. For these reasons, Occupational Safety and Health Administration in the United States, and similar institutions in other developed countries require personnel working with 32
P to wear lab coats, disposable gloves, and safety glasses or goggles to protect the eyes, and avoid working directly over open containers. Monitoring personal, clothing, and surface contamination is also required. Shielding requires special consideration. The high energy of the beta particles gives rise to secondary emission of X-rays via Bremsstrahlung (braking radiation) in dense shielding materials such as lead. Therefore, the radiation must be shielded with low density materials such as acrylic or other plastic, water, or (when transparency is not required), even wood.[34]
Occurrence[edit]
Universe[edit]
In 2013, astronomers detected phosphorus in Cassiopeia A, which confirmed that this element is produced in supernovae as a byproduct of supernova nucleosynthesis. The phosphorus-to-iron ratio in material from the supernova remnant could be up to 100 times higher than in the Milky Way in general.[35]
In 2020, astronomers analysed ALMA and ROSINA data from the massive star-forming region AFGL 5142, to detect phosphorus-bearing molecules and how they are carried in comets to the early Earth.[36][37]
Crust and organic sources[edit]
Phosphorus has a concentration in the Earth’s crust of about one gram per kilogram (compare copper at about 0.06 grams). It is not found free in nature, but is widely distributed in many minerals, usually as phosphates.[9] Inorganic phosphate rock, which is partially made of apatite (a group of minerals being, generally, pentacalcium triorthophosphate fluoride (hydroxide)), is today the chief commercial source of this element. According to the US Geological Survey (USGS), about 50 percent of the global phosphorus reserves are in the Arab nations.[38] 85% of Earth’s known reserves are in Morocco with smaller deposits in China, Russia,[39] Florida, Idaho, Tennessee, Utah, and elsewhere.[40] Albright and Wilson in the UK and their Niagara Falls plant, for instance, were using phosphate rock in the 1890s and 1900s from Tennessee, Florida, and the Îles du Connétable (guano island sources of phosphate); by 1950, they were using phosphate rock mainly from Tennessee and North Africa.[41]
Organic sources, namely urine, bone ash and (in the latter 19th century) guano, were historically of importance but had only limited commercial success.[42] As urine contains phosphorus, it has fertilising qualities which are still harnessed today in some countries, including Sweden, using methods for reuse of excreta. To this end, urine can be used as a fertiliser in its pure form or part of being mixed with water in the form of sewage or sewage sludge.
Compounds[edit]
Phosphorus(V)[edit]

The tetrahedral structure of P4O10 and P4S10.
The most prevalent compounds of phosphorus are derivatives of phosphate (PO43−), a tetrahedral anion.[43] Phosphate is the conjugate base of phosphoric acid, which is produced on a massive scale for use in fertilisers. Being triprotic, phosphoric acid converts stepwise to three conjugate bases:
- H3PO4 + H2O ⇌ H3O+ + H2PO4− Ka1 = 7.25×10−3
- H2PO4− + H2O ⇌ H3O+ + HPO42− Ka2 = 6.31×10−8
- HPO42− + H2O ⇌ H3O+ + PO43− Ka3 = 3.98×10−13
Phosphate exhibits a tendency to form chains and rings containing P-O-P bonds. Many polyphosphates are known, including ATP. Polyphosphates arise by dehydration of hydrogen phosphates such as HPO42− and H2PO4−. For example, the industrially important pentasodium triphosphate (also known as sodium tripolyphosphate, STPP) is produced industrially on by the megatonne by this condensation reaction:
- 2 Na2[(HO)PO3] + Na[(HO)2PO2] → Na5[O3P-O-P(O)2-O-PO3] + 2 H2O
Phosphorus pentoxide (P4O10) is the acid anhydride of phosphoric acid, but several intermediates between the two are known. This waxy white solid reacts vigorously with water.
With metal cations, phosphate forms a variety of salts. These solids are polymeric, featuring P-O-M linkages. When the metal cation has a charge of 2+ or 3+, the salts are generally insoluble, hence they exist as common minerals. Many phosphate salts are derived from hydrogen phosphate (HPO42−).
PCl5 and PF5 are common compounds. PF5 is a colourless gas and the molecules have trigonal bipyramidal geometry. PCl5 is a colourless solid which has an ionic formulation of PCl4+ PCl6−, but adopts the trigonal bipyramidal geometry when molten or in the vapour phase.[15] PBr5 is an unstable solid formulated as PBr4+Br−and PI5 is not known.[15] The pentachloride and pentafluoride are Lewis acids. With fluoride, PF5 forms PF6−, an anion that is isoelectronic with SF6. The most important oxyhalide is phosphorus oxychloride, (POCl3), which is approximately tetrahedral.
Before extensive computer calculations were feasible, it was thought that bonding in phosphorus(V) compounds involved d orbitals. Computer modeling of molecular orbital theory indicates that this bonding involves only s- and p-orbitals.[44]
Phosphorus(III)[edit]
All four symmetrical trihalides are well known: gaseous PF3, the yellowish liquids PCl3 and PBr3, and the solid PI3. These materials are moisture sensitive, hydrolysing to give phosphorous acid. The trichloride, a common reagent, is produced by chlorination of white phosphorus:
- P4 + 6 Cl2 → 4 PCl3
The trifluoride is produced from the trichloride by halide exchange. PF3 is toxic because it binds to haemoglobin.
Phosphorus(III) oxide, P4O6 (also called tetraphosphorus hexoxide) is the anhydride of P(OH)3, the minor tautomer of phosphorous acid. The structure of P4O6 is like that of P4O10 without the terminal oxide groups.
Phosphorus(I) and phosphorus(II)[edit]

These compounds generally feature P–P bonds.[15] Examples include catenated derivatives of phosphine and organophosphines. Compounds containing P=P double bonds have also been observed, although they are rare.
Phosphides and phosphines[edit]
Phosphides arise by reaction of metals with red phosphorus. The alkali metals (group 1) and alkaline earth metals can form ionic compounds containing the phosphide ion, P3−. These compounds react with water to form phosphine. Other phosphides, for example Na3P7, are known for these reactive metals. With the transition metals as well as the monophosphides there are metal-rich phosphides, which are generally hard refractory compounds with a metallic lustre, and phosphorus-rich phosphides which are less stable and include semiconductors.[15] Schreibersite is a naturally occurring metal-rich phosphide found in meteorites. The structures of the metal-rich and phosphorus-rich phosphides can be complex.
Phosphine (PH3) and its organic derivatives (PR3) are structural analogues of ammonia (NH3), but the bond angles at phosphorus are closer to 90° for phosphine and its organic derivatives. It is an ill-smelling, toxic compound. Phosphorus has an oxidation number of −3 in phosphine. Phosphine is produced by hydrolysis of calcium phosphide, Ca3P2. Unlike ammonia, phosphine is oxidised by air. Phosphine is also far less basic than ammonia. Other phosphines are known which contain chains of up to nine phosphorus atoms and have the formula PnHn+2.[15] The highly flammable gas diphosphine (P2H4) is an analogue of hydrazine.
Oxoacids[edit]
Phosphorous oxoacids are extensive, often commercially important, and sometimes structurally complicated. They all have acidic protons bound to oxygen atoms, some have nonacidic protons that are bonded directly to phosphorus and some contain phosphorus — phosphorus bonds.[15] Although many oxoacids of phosphorus are formed, only nine are commercially important, and three of them, hypophosphorous acid, phosphorous acid, and phosphoric acid, are particularly important.
| Oxidation state | Formula | Name | Acidic protons | Compounds |
|---|---|---|---|---|
| +1 | HH2PO2 | hypophosphorous acid | 1 | acid, salts |
| +3 | H2HPO3 | phosphorous acid | 2 | acid, salts |
| +3 | HPO2 | metaphosphorous acid | 1 | salts |
| +3 | H3PO3 | (ortho)phosphorous acid | 3 | acid, salts |
| +4 | H4P2O6 | hypophosphoric acid | 4 | acid, salts |
| +5 | (HPO3)n | metaphosphoric acids | n | salts (n = 3,4,6) |
| +5 | H(HPO3)nOH | polyphosphoric acids | n+2 | acids, salts (n = 1-6) |
| +5 | H5P3O10 | tripolyphosphoric acid | 3 | salts |
| +5 | H4P2O7 | pyrophosphoric acid | 4 | acid, salts |
| +5 | H3PO4 | (ortho)phosphoric acid | 3 | acid, salts |
Nitrides[edit]
The PN molecule is considered unstable, but is a product of crystalline phosphorus nitride decomposition at 1100 K. Similarly, H2PN is considered unstable, and phosphorus nitride halogens like F2PN, Cl2PN, Br2PN, and I2PN oligomerise into cyclic Polyphosphazenes. For example, compounds of the formula (PNCl2)n exist mainly as rings such as the trimer hexachlorophosphazene. The phosphazenes arise by treatment of phosphorus pentachloride with ammonium chloride:
PCl5 + NH4Cl → 1/n (NPCl2)n + 4 HCl
When the chloride groups are replaced by alkoxide (RO−), a family of polymers is produced with potentially useful properties.[45]
Sulfides[edit]
Phosphorus forms a wide range of sulfides, where the phosphorus can be in P(V), P(III) or other oxidation states. The three-fold symmetric P4S3 is used in strike-anywhere matches. P4S10 and P4O10 have analogous structures.[46] Mixed oxyhalides and oxyhydrides of phosphorus(III) are almost unknown.
Organophosphorus compounds[edit]
Compounds with P-C and P-O-C bonds are often classified as organophosphorus compounds. They are widely used commercially. The PCl3 serves as a source of P3+ in routes to organophosphorus(III) compounds. For example, it is the precursor to triphenylphosphine:
- PCl3 + 6 Na + 3 C6H5Cl → P(C6H5)3 + 6 NaCl
Treatment of phosphorus trihalides with alcohols and phenols gives phosphites, e.g. triphenylphosphite:
- PCl3 + 3 C6H5OH → P(OC6H5)3 + 3 HCl
Similar reactions occur for phosphorus oxychloride, affording triphenylphosphate:
- OPCl3 + 3 C6H5OH → OP(OC6H5)3 + 3 HCl
History[edit]
Etymology[edit]
The name Phosphorus in Ancient Greece was the name for the planet Venus and is derived from the Greek words (φῶς = light, φέρω = carry), which roughly translates as light-bringer or light carrier.[19] (In Greek mythology and tradition, Augerinus (Αυγερινός = morning star, still in use today), Hesperus or Hesperinus (΄Εσπερος or Εσπερινός or Αποσπερίτης = evening star, still in use today) and Eosphorus (Εωσφόρος = dawnbearer, not in use for the planet after Christianity) are close homologues, and also associated with Phosphorus-the-morning-star).
According to the Oxford English Dictionary, the correct spelling of the element is phosphorus. The word phosphorous is the adjectival form of the P3+ valence: so, just as sulfur forms sulfurous and sulfuric compounds, phosphorus forms phosphorous compounds (e.g., phosphorous acid) and P5+ valence phosphoric compounds (e.g., phosphoric acids and phosphates).
Discovery[edit]

The discovery of phosphorus, the first element to be discovered that was not known since ancient times,[47] is credited to the German alchemist Hennig Brand in 1669, although others might have discovered phosphorus around the same time.[48] Brand experimented with urine, which contains considerable quantities of dissolved phosphates from normal metabolism.[19] Working in Hamburg, Brand attempted to create the fabled philosopher’s stone through the distillation of some salts by evaporating urine, and in the process produced a white material that glowed in the dark and burned brilliantly. It was named phosphorus mirabilis («miraculous bearer of light»).[49]
Brand’s process originally involved letting urine stand for days until it gave off a terrible smell. Then he boiled it down to a paste, heated this paste to a high temperature, and led the vapours through water, where he hoped they would condense to gold. Instead, he obtained a white, waxy substance that glowed in the dark. Brand had discovered phosphorus. Specifically, Brand produced ammonium sodium hydrogen phosphate, (NH
4)NaHPO
4. While the quantities were essentially correct (it took about 1,100 litres [290 US gal] of urine to make about 60 g of phosphorus), it was unnecessary to allow the urine to rot first. Later scientists discovered that fresh urine yielded the same amount of phosphorus.[31]
Brand at first tried to keep the method secret,[50] but later sold the recipe for 200 thalers to D. Krafft from Dresden.[19] Krafft toured much of Europe with it, including England, where he met with Robert Boyle. The secret—that the substance was made from urine—leaked out, and Johann Kunckel (1630–1703) was able to reproduce it in Sweden (1678). Later, Boyle in London (1680) also managed to make phosphorus, possibly with the aid of his assistant, Ambrose Godfrey-Hanckwitz. Godfrey later made a business of the manufacture of phosphorus.
Boyle states that Krafft gave him no information as to the preparation of phosphorus other than that it was derived from «somewhat that belonged to the body of man». This gave Boyle a valuable clue, so that he, too, managed to make phosphorus, and published the method of its manufacture.[19] Later he improved Brand’s process by using sand in the reaction (still using urine as base material),
- 4 NaPO
3 + 2 SiO
2 + 10 C → 2 Na
2SiO
3 + 10 CO + P
4
Robert Boyle was the first to use phosphorus to ignite sulfur-tipped wooden splints, forerunners of our modern matches, in 1680.[51]
Phosphorus was the 13th element to be discovered. Because of its tendency to spontaneously combust when left alone in air, it is sometimes referred to as «the Devil’s element».[52]
Bone ash and guano[edit]

Antoine Lavoisier recognized phosphorus as an element in 1777 after Johan Gottlieb Gahn and Carl Wilhelm Scheele, in 1769, showed that calcium phosphate (Ca
3(PO
4)
2) is found in bones by obtaining elemental phosphorus from bone ash.[53]
Bone ash was the major source of phosphorus until the 1840s. The method started by roasting bones, then employed the use of fire clay retorts encased in a very hot brick furnace to distill out the highly toxic elemental phosphorus product.[54] Alternately, precipitated phosphates could be made from ground-up bones that had been de-greased and treated with strong acids. White phosphorus could then be made by heating the precipitated phosphates, mixed with ground coal or charcoal in an iron pot, and distilling off phosphorus vapour in a retort.[55] Carbon monoxide and other flammable gases produced during the reduction process were burnt off in a flare stack.
In the 1840s, world phosphate production turned to the mining of tropical island deposits formed from bird and bat guano (see also Guano Islands Act). These became an important source of phosphates for fertiliser in the latter half of the 19th century.[56]
Phosphate rock[edit]
Phosphate rock, which usually contains calcium phosphate, was first used in 1850 to make phosphorus, and following the introduction of the electric arc furnace by James Burgess Readman in 1888[57] (patented 1889),[58] elemental phosphorus production switched from the bone-ash heating, to electric arc production from phosphate rock. After the depletion of world guano sources about the same time, mineral phosphates became the major source of phosphate fertiliser production. Phosphate rock production greatly increased after World War II, and remains the primary global source of phosphorus and phosphorus chemicals today. See the article on peak phosphorus for more information on the history and present state of phosphate mining. Phosphate rock remains a feedstock in the fertiliser industry, where it is treated with sulfuric acid to produce various «superphosphate» fertiliser products.
Incendiaries[edit]
White phosphorus was first made commercially in the 19th century for the match industry. This used bone ash for a phosphate source, as described above. The bone-ash process became obsolete when the submerged-arc furnace for phosphorus production was introduced to reduce phosphate rock.[59][60] The electric furnace method allowed production to increase to the point where phosphorus could be used in weapons of war.[29][61] In World War I, it was used in incendiaries, smoke screens and tracer bullets.[61] A special incendiary bullet was developed to shoot at hydrogen-filled Zeppelins over Britain (hydrogen being highly flammable).[61] During World War II, Molotov cocktails made of phosphorus dissolved in petrol were distributed in Britain to specially selected civilians within the British resistance operation, for defence; and phosphorus incendiary bombs were used in war on a large scale. Burning phosphorus is difficult to extinguish and if it splashes onto human skin it has horrific effects.[15]
Early matches used white phosphorus in their composition, which was dangerous due to its toxicity. Murders, suicides and accidental poisonings resulted from its use. (An apocryphal tale tells of a woman attempting to murder her husband with white phosphorus in his food, which was detected by the stew’s giving off luminous steam).[29] In addition, exposure to the vapours gave match workers a severe necrosis of the bones of the jaw, known as «phossy jaw». When a safe process for manufacturing red phosphorus was discovered, with its far lower flammability and toxicity, laws were enacted, under the Berne Convention (1906), requiring its adoption as a safer alternative for match manufacture.[62] The toxicity of white phosphorus led to discontinuation of its use in matches.[63] The Allies used phosphorus incendiary bombs in World War II to destroy Hamburg, the place where the «miraculous bearer of light» was first discovered.[49]
Production[edit]

Mining of phosphate rock in Nauru
In 2017, the USGS estimated 68 billion tons of world reserves, where reserve figures refer to the amount assumed recoverable at current market prices; 0.261 billion tons were mined in 2016.[64] Critical to contemporary agriculture, its annual demand is rising nearly twice as fast as the growth of the human population.[39] The production of phosphorus may have peaked before 2011 and some scientists predict reserves will be depleted in before the end of the 21st Century.»[65][39][66] Phosphorus comprises about 0.1% by mass of the average rock, and consequently, the Earth’s supply is vast, though dilute.[15]
Wet process[edit]
Most phosphorus-bearing material is for agriculture fertilisers. In this case where the standards of purity are modest, phosphorus is obtained from phosphate rock by what is called the «wet process.» The minerals are treated with sulfuric acid to give phosphoric acid. Phosphoric acid is then neutralized to give various phosphate salts, which comprise fertilizers. In the wet process, phosphorus does not undergo redox.[67] About five tons of phosphogypsum waste are generated per ton of phosphoric acid production. Annually, the estimated generation of phosphogypsum worldwide is 100 to 280 Mt.[68]
Thermal process[edit]
For the use of phosphorus in drugs, detergents, and foodstuff, the standards of purity are high, which led to the development of the thermal process. In this process, phosphate minerals are converted to white phosphorus, which can be purified by distillation. The white phosphorus is then oxidised to phosphoric acid and subsequently neutralised with a base to give phosphate salts. The thermal process is energy intensive.[67] Presently, about 1,000,000 short tons (910,000 t) of elemental phosphorus is produced annually. Calcium phosphate (phosphate rock), mostly mined in Florida and North Africa, can be heated to 1,200–1,500 °C with sand, which is mostly SiO
2, and coke to produce P
4. The P
4 product, being volatile, is readily isolated:[69]
- 4 Ca5(PO4)3F + 18 SiO2 + 30 C → 3 P4 + 30 CO + 18 CaSiO3 + 2 CaF2
- 2 Ca3(PO4)2 + 6 SiO2 + 10 C → 6 CaSiO3 + 10 CO + P4
Side products from the thermal process include ferrophosphorus, a crude form of Fe2P, resulting from iron impurities in the mineral precursors. The silicate slag is a useful construction material. The fluoride is sometimes recovered for use in water fluoridation. More problematic is a «mud» containing significant amounts of white phosphorus. Production of white phosphorus is conducted in large facilities in part because it is energy intensive. The white phosphorus is transported in molten form. Some major accidents have occurred during transportation.[70]
Historical routes[edit]
Historically, before the development of mineral-based extractions, white phosphorus was isolated on an industrial scale from bone ash.[71] In this process, the tricalcium phosphate in bone ash is converted to monocalcium phosphate with sulfuric acid:
- Ca3(PO4)2 + 2 H2SO4 → Ca(H2PO4)2 + 2 CaSO4
Monocalcium phosphate is then dehydrated to the corresponding metaphosphate:
- Ca(H2PO4)2 → Ca(PO3)2 + 2 H2O
When ignited to a white heat (~1300C) with charcoal, calcium metaphosphate yields two-thirds of its weight of white phosphorus while one-third of the phosphorus remains in the residue as calcium orthophosphate:
- 3 Ca(PO3)2 + 10 C → Ca3(PO4)2 + 10 CO + P4
Applications[edit]
Food additive[edit]
Phosphorus is an essential mineral for humans listed in the Dietary Reference Intake (DRI).
Food-grade phosphoric acid (additive E338[72]) is used to acidify foods and beverages such as various colas and jams, providing a tangy or sour taste. The phosphoric acid also serves as a preservative.[73] Soft drinks containing phosphoric acid, which would include Coca-Cola, are sometimes called phosphate sodas or phosphates. Phosphoric acid in soft drinks has the potential to cause dental erosion.[74] Phosphoric acid also has the potential to contribute to the formation of kidney stones, especially in those who have had kidney stones previously.[75]
Fertiliser[edit]
Phosphorus is an essential plant nutrient (the most often limiting nutrient, after nitrogen),[76] and the bulk of all phosphorus production is in concentrated phosphoric acids for agriculture fertilisers, containing as much as 70% to 75% P2O5. That led to large increase in phosphate (PO43−) production in the second half of the 20th century.[39] Artificial phosphate fertilisation is necessary because phosphorus is essential to all living organisms; it is involved in energy transfers, strength of root and stems, photosynthesis, the expansion of plant roots, formation of seeds and flowers, and other important factors effecting overall plant health and genetics.[76]
Natural phosphorus-bearing compounds are mostly inaccessible to plants because of the low solubility and mobility in soil.[77] Most phosphorus is very stable in the soil minerals or organic matter of the soil. Even when phosphorus is added in manure or fertilizer it can become fixed in the soil. Therefore, the natural cycle of phosphorus is very slow. Some of the fixed phosphorus is released again over time, sustaining wild plant growth, however, more is needed to sustain intensive cultivation of crops.[78] Fertiliser is often in the form of superphosphate of lime, a mixture of calcium dihydrogen phosphate (Ca(H2PO4)2), and calcium sulfate dihydrate (CaSO4·2H2O) produced reacting sulfuric acid and water with calcium phosphate.
Processing phosphate minerals with sulfuric acid for obtaining fertiliser is so important to the global economy that this is the primary industrial market for sulfuric acid and the greatest industrial use of elemental sulfur.[79]
| Widely used compounds | Use |
|---|---|
| Ca(H2PO4)2·H2O | Baking powder and fertilisers |
| CaHPO4·2H2O | Animal food additive, toothpowder |
| H3PO4 | Manufacture of phosphate fertilisers |
| PCl3 | Manufacture of POCl3 and pesticides |
| POCl3 | Manufacture of plasticiser |
| P4S10 | Manufacturing of additives and pesticides |
| Na5P3O10 | Detergents |
Organophosphorus[edit]
White phosphorus is widely used to make organophosphorus compounds through intermediate phosphorus chlorides and two phosphorus sulfides, phosphorus pentasulfide and phosphorus sesquisulfide.[80] Organophosphorus compounds have many applications, including in plasticisers, flame retardants, pesticides, extraction agents, nerve agents and water treatment.[15][81]
Metallurgical aspects[edit]
Phosphorus is also an important component in steel production, in the making of phosphor bronze, and in many other related products.[82][83] Phosphorus is added to metallic copper during its smelting process to react with oxygen present as an impurity in copper and to produce phosphorus-containing copper (CuOFP) alloys with a higher hydrogen embrittlement resistance than normal copper.[84]
Matches[edit]

Match striking surface made of a mixture of red phosphorus, glue and ground glass. The glass powder is used to increase the friction.
The first striking match with a phosphorus head was invented by Charles Sauria in 1830. These matches (and subsequent modifications) were made with heads of white phosphorus, an oxygen-releasing compound (potassium chlorate, lead dioxide, or sometimes nitrate), and a binder. They were poisonous to the workers in manufacture,[85] sensitive to storage conditions, toxic if ingested, and hazardous when accidentally ignited on a rough surface.[86][87] Production in several countries was banned between 1872 and 1925.[88] The international Berne Convention, ratified in 1906, prohibited the use of white phosphorus in matches.
In consequence, phosphorous matches were gradually replaced by safer alternatives. Around 1900 French chemists Henri Sévène and Emile David Cahen invented the modern strike-anywhere match, wherein the white phosphorus was replaced by phosphorus sesquisulfide (P4S3), a non-toxic and non-pyrophoric compound that ignites under friction. For a time these safer strike-anywhere matches were quite popular but in the long run they were superseded by the modern safety match.
Safety matches are very difficult to ignite on any surface other than a special striker strip. The strip contains non-toxic red phosphorus and the match head potassium chlorate, an oxygen-releasing compound. When struck, small amounts of abrasion from match head and striker strip are mixed intimately to make a small quantity of Armstrong’s mixture, a very touch sensitive composition. The fine powder ignites immediately and provides the initial spark to set off the match head. Safety matches separate the two components of the ignition mixture until the match is struck. This is the key safety advantage as it prevents accidental ignition. Nonetheless, safety matches, invented in 1844 by Gustaf Erik Pasch and market ready by the 1860s, didn’t gain consumer acceptance until the prohibition of white phosphorus. Using a dedicated striker strip was considered clumsy.[20][80][89]
Water softening[edit]
Sodium tripolyphosphate made from phosphoric acid is used in laundry detergents in some countries, but banned for this use in others.[22] This compound softens the water to enhance the performance of the detergents and to prevent pipe/boiler tube corrosion.[90]
Miscellaneous[edit]
- Phosphates are used to make special glasses for sodium lamps.[22]
- Bone-ash, calcium phosphate, is used in the production of fine china.[22]
- Phosphoric acid made from elemental phosphorus is used in food applications such as soft drinks, and as a starting point for food grade phosphates.[80] These include mono-calcium phosphate for baking powder and sodium tripolyphosphate.[80] Phosphates are used to improve the characteristics of processed meat and cheese, and in toothpaste.[80]
- White phosphorus, called «WP» (slang term «Willie Peter») is used in military applications as incendiary bombs, for smoke-screening as smoke pots and smoke bombs, and in tracer ammunition. It is also a part of an obsolete M34 White Phosphorus US hand grenade. This multipurpose grenade was mostly used for signaling, smoke screens, and inflammation; it could also cause severe burns and had a psychological impact on the enemy.[91][92] Military uses of white phosphorus are constrained by international law.
- 32P and 33P are used as radioactive tracers in biochemical laboratories.[93]
Biological role[edit]
Inorganic phosphorus in the form of the phosphate PO3−
4 is required for all known forms of life.[94] Phosphorus plays a major role in the structural framework of DNA and RNA. Living cells use phosphate to transport cellular energy with adenosine triphosphate (ATP), necessary for every cellular process that uses energy. ATP is also important for phosphorylation, a key regulatory event in cells. Phospholipids are the main structural components of all cellular membranes. Calcium phosphate salts assist in stiffening bones.[15] Biochemists commonly use the abbreviation «Pi» to refer to inorganic phosphate.[95]
Every living cell is encased in a membrane that separates it from its surroundings. Cellular membranes are composed of a phospholipid matrix and proteins, typically in the form of a bilayer. Phospholipids are derived from glycerol with two of the glycerol hydroxyl (OH) protons replaced by fatty acids as an ester, and the third hydroxyl proton has been replaced with phosphate bonded to another alcohol.[96]
An average adult human contains about 0.7 kg of phosphorus, about 85–90% in bones and teeth in the form of apatite, and the remainder in soft tissues and extracellular fluids (~1%). The phosphorus content increases from about 0.5% by mass in infancy to 0.65–1.1% by mass in adults. Average phosphorus concentration in the blood is about 0.4 g/L, about 70% of that is organic and 30% inorganic phosphates.[97] An adult with healthy diet consumes and excretes about 1–3 grams of phosphorus per day, with consumption in the form of inorganic phosphate and phosphorus-containing biomolecules such as nucleic acids and phospholipids; and excretion almost exclusively in the form of phosphate ions such as H
2PO−
4 and HPO2−
4. Only about 0.1% of body phosphate circulates in the blood, paralleling the amount of phosphate available to soft tissue cells.
Bone and teeth enamel[edit]
The main component of bone is hydroxyapatite as well as amorphous forms of calcium phosphate, possibly including carbonate. Hydroxyapatite is the main component of tooth enamel. Water fluoridation enhances the resistance of teeth to decay by the partial conversion of this mineral to the still harder material called fluoroapatite:[15]
- Ca
5(PO
4)
3OH + F−
→ Ca
5(PO
4)
3F + OH−
Phosphorus deficiency[edit]
In medicine, phosphate deficiency syndrome may be caused by malnutrition, by failure to absorb phosphate, and by metabolic syndromes that draw phosphate from the blood (such as in refeeding syndrome after malnutrition[98]) or passing too much of it into the urine. All are characterised by hypophosphatemia, which is a condition of low levels of soluble phosphate levels in the blood serum and inside the cells. Symptoms of hypophosphatemia include neurological dysfunction and disruption of muscle and blood cells due to lack of ATP. Too much phosphate can lead to diarrhoea and calcification (hardening) of organs and soft tissue, and can interfere with the body’s ability to use iron, calcium, magnesium, and zinc.[99]
Phosphorus is an essential macromineral for plants, which is studied extensively in edaphology to understand plant uptake from soil systems. Phosphorus is a limiting factor in many ecosystems; that is, the scarcity of phosphorus limits the rate of organism growth. An excess of phosphorus can also be problematic, especially in aquatic systems where eutrophication sometimes leads to algal blooms.[39]
Nutrition[edit]
Dietary recommendations[edit]
The U.S. Institute of Medicine (IOM) updated Estimated Average Requirements (EARs) and Recommended Dietary Allowances (RDAs) for phosphorus in 1997. If there is not sufficient information to establish EARs and RDAs, an estimate designated Adequate Intake (AI) is used instead. The current EAR for phosphorus for people ages 19 and up is 580 mg/day. The RDA is 700 mg/day. RDAs are higher than EARs so as to identify amounts that will cover people with higher than average requirements. RDA for pregnancy and lactation are also 700 mg/day. For people ages 1–18 years the RDA increases with age from 460 to 1250 mg/day. As for safety, the IOM sets Tolerable upper intake levels (ULs) for vitamins and minerals when evidence is sufficient. In the case of phosphorus the UL is 4000 mg/day. Collectively the EARs, RDAs, AIs and ULs are referred to as Dietary Reference Intakes (DRIs).[100]
The European Food Safety Authority (EFSA) refers to the collective set of information as Dietary Reference Values, with Population Reference Intake (PRI) instead of RDA, and Average Requirement instead of EAR. AI and UL defined the same as in United States. For people ages 15 and older, including pregnancy and lactation, the AI is set at 550 mg/day. For children ages 4–10 the AI is 440 mg/day, and for ages 11–17 it is 640 mg/day. These AIs are lower than the U.S RDAs. In both systems, teenagers need more than adults.[101] The European Food Safety Authority reviewed the same safety question and decided that there was not sufficient information to set a UL.[102]
For U.S. food and dietary supplement labeling purposes the amount in a serving is expressed as a percent of Daily Value (%DV). For phosphorus labeling purposes 100% of the Daily Value was 1000 mg, but as of May 27, 2016 it was revised to 1250 mg to bring it into agreement with the RDA.[103][104] A table of the old and new adult daily values is provided at Reference Daily Intake.
Food sources[edit]
The main food sources for phosphorus are the same as those containing protein, although proteins do not contain phosphorus. For example, milk, meat, and soya typically also have phosphorus. As a rule, if a diet has sufficient protein and calcium, the amount of phosphorus is probably sufficient.[105]
Precautions[edit]

Organic compounds of phosphorus form a wide class of materials; many are required for life, but some are extremely toxic. Fluorophosphate esters are among the most potent neurotoxins known. A wide range of organophosphorus compounds are used for their toxicity as pesticides (herbicides, insecticides, fungicides, etc.) and weaponised as nerve agents against enemy humans. Most inorganic phosphates are relatively nontoxic and essential nutrients.[15]
The white phosphorus allotrope presents a significant hazard because it ignites in air and produces phosphoric acid residue. Chronic white phosphorus poisoning leads to necrosis of the jaw called «phossy jaw». White phosphorus is toxic, causing severe liver damage on ingestion and may cause a condition known as «Smoking Stool Syndrome».[106]
In the past, external exposure to elemental phosphorus was treated by washing the affected area with 2% copper sulfate solution to form harmless compounds that are then washed away. According to the recent US Navy’s Treatment of Chemical Agent Casualties and Conventional Military Chemical Injuries: FM8-285: Part 2 Conventional Military Chemical Injuries, «Cupric (copper(II)) sulfate has been used by U.S. personnel in the past and is still being used by some nations. However, copper sulfate is toxic and its use will be discontinued. Copper sulfate may produce kidney and cerebral toxicity as well as intravascular hemolysis.»[107]
The manual suggests instead «a bicarbonate solution to neutralise phosphoric acid, which will then allow removal of visible white phosphorus. Particles often can be located by their emission of smoke when air strikes them, or by their phosphorescence in the dark. In dark surroundings, fragments are seen as luminescent spots. Promptly debride the burn if the patient’s condition will permit removal of bits of WP (white phosphorus) that might be absorbed later and possibly produce systemic poisoning. DO NOT apply oily-based ointments until it is certain that all WP has been removed. Following complete removal of the particles, treat the lesions as thermal burns.»[note 1][citation needed] As white phosphorus readily mixes with oils, any oily substances or ointments are not recommended until the area is thoroughly cleaned and all white phosphorus removed.
People can be exposed to phosphorus in the workplace by inhalation, ingestion, skin contact, and eye contact. The Occupational Safety and Health Administration (OSHA) has set the phosphorus exposure limit (Permissible exposure limit) in the workplace at 0.1 mg/m3 over an 8-hour workday. The National Institute for Occupational Safety and Health (NIOSH) has set a Recommended exposure limit (REL) of 0.1 mg/m3 over an 8-hour workday. At levels of 5 mg/m3, phosphorus is immediately dangerous to life and health.[108]
US DEA List I status[edit]
Phosphorus can reduce elemental iodine to hydroiodic acid, which is a reagent effective for reducing ephedrine or pseudoephedrine to methamphetamine.[109] For this reason, red and white phosphorus were designated by the United States Drug Enforcement Administration as List I precursor chemicals under 21 CFR 1310.02 effective on November 17, 2001.[110] In the United States, handlers of red or white phosphorus are subject to stringent regulatory controls.[110][111][112]
See also[edit]
- Phosphorus cycle
Notes[edit]
- ^ WP, (white phosphorus), exhibits chemoluminescence upon exposure to air and if there is any WP in the wound, covered by tissue or fluids such as blood serum, it will not glow until it is exposed to air, which requires a very dark room and dark-adapted eyes to see clearly
References[edit]
- ^ «Standard Atomic Weights: Phosphorus». CIAAW. 2013.
- ^ «Phosphorus: Chemical Element». Encyclopædia Britannica.
- ^ Wang, Yuzhong; Xie, Yaoming; Wei, Pingrong; King, R. Bruce; Schaefer, Iii; Schleyer, Paul v. R.; Robinson, Gregory H. (2008). «Carbene-Stabilized Diphosphorus». Journal of the American Chemical Society. 130 (45): 14970–1. doi:10.1021/ja807828t. PMID 18937460.
- ^ Ellis, Bobby D.; MacDonald, Charles L. B. (2006). «Phosphorus(I) Iodide: A Versatile Metathesis Reagent for the Synthesis of Low Oxidation State Phosphorus Compounds». Inorganic Chemistry. 45 (17): 6864–74. doi:10.1021/ic060186o. PMID 16903744.
- ^ Lide, D. R., ed. (2005). «Magnetic susceptibility of the elements and inorganic compounds». CRC Handbook of Chemistry and Physics (PDF) (86th ed.). Boca Raton (FL): CRC Press. ISBN 0-8493-0486-5.
- ^ Weast, Robert (1984). CRC, Handbook of Chemistry and Physics. Boca Raton, Florida: Chemical Rubber Company Publishing. pp. E110. ISBN 0-8493-0464-4.
- ^ cf. «Memoir on Combustion in General» Mémoires de l’Académie Royale des Sciences 1777, 592–600. from Henry Marshall Leicester and Herbert S. Klickstein, A Source Book in Chemistry 1400–1900 (New York: McGraw Hill, 1952)
- ^ a b A. Holleman; N. Wiberg (1985). «XV 2.1.3». Lehrbuch der Anorganischen Chemie (33rd ed.). de Gruyter. ISBN 3-11-012641-9.
- ^ a b Abundance. ptable.com
- ^ Simon, Arndt; Borrmann, Horst; Horakh, Jörg (1997). «On the Polymorphism of White Phosphorus». Chemische Berichte. 130 (9): 1235–1240. doi:10.1002/cber.19971300911.
- ^ a b Cossairt, Brandi M.; Cummins, Christopher C.; Head, Ashley R.; Lichtenberger, Dennis L.; Berger, Raphael J. F.; Hayes, Stuart A.; Mitzel, Norbert W.; Wu, Gang (2010-06-01). «On the Molecular and Electronic Structures of AsP3 and P4». Journal of the American Chemical Society. 132 (24): 8459–8465. doi:10.1021/ja102580d. ISSN 0002-7863. PMID 20515032.
- ^ Welford C. Roberts; William R. Hartley (1992-06-16). Drinking Water Health Advisory: Munitions (illustrated ed.). CRC Press, 1992. p. 399. ISBN 0873717546.
- ^ Marie-Thérèse Averbuch-Pouchot; A. Durif (1996). Topics in Phosphate Chemistry. World Scientific, 1996. p. 3. ISBN 9810226349.
- ^ Simon, Arndt; Borrmann, Horst; Horakh, Jörg (September 1997). «On the Polymorphism of White Phosphorus». Chemische Berichte. 130 (9): 1235–1240. doi:10.1002/cber.19971300911. ISSN 0009-2940.
- ^ a b c d e f g h i j k l m n Greenwood, N. N.; & Earnshaw, A. (1997). Chemistry of the Elements (2nd Edn.), Oxford:Butterworth-Heinemann. ISBN 0-7506-3365-4.
- ^ Piro, N. A.; Figueroa, J. S.; McKellar, J. T.; Cummins, C. C. (2006). «Triple-Bond Reactivity of Diphosphorus Molecules». Science. 313 (5791): 1276–9. Bibcode:2006Sci…313.1276P. doi:10.1126/science.1129630. PMID 16946068. S2CID 27740669.
- ^ a b c Berger, L. I. (1996). Semiconductor materials. CRC Press. p. 84. ISBN 0-8493-8912-7.
- ^ Shen, Z; Yu, JC (2016). «Nanostructured elemental photocatalysts: Development and challenges». In Yamashita, H; Li, H (eds.). Nanostructured Photocatalysts: Advanced Functional Materials. Switzerland: Springer. pp. 295–312 (301). ISBN 978-3-319-26077-8.
- ^ a b c d e Parkes & Mellor 1939, p. 717
- ^ a b Egon Wiberg; Nils Wiberg; Arnold Frederick Holleman (2001). Inorganic chemistry. Academic Press. pp. 683–684, 689. ISBN 978-0-12-352651-9. Retrieved 2011-11-19.
- ^ Parkes & Mellor 1939, pp. 721–722
- ^ a b c d Hammond, C. R. (2000). The Elements, in Handbook of Chemistry and Physics (81st ed.). CRC press. ISBN 0-8493-0481-4.
- ^ A. Brown; S. Runquist (1965). «Refinement of the crystal structure of black phosphorus». Acta Crystallogr. 19 (4): 684–685. doi:10.1107/S0365110X65004140.
- ^ Cartz, L.; Srinivasa, S.R.; Riedner, R.J.; Jorgensen, J.D.; Worlton, T.G. (1979). «Effect of pressure on bonding in black phosphorus». Journal of Chemical Physics. 71 (4): 1718–1721. Bibcode:1979JChPh..71.1718C. doi:10.1063/1.438523.
- ^ Lange, Stefan; Schmidt, Peer & Nilges, Tom (2007). «Au3SnP7@Black Phosphorus: An Easy Access to Black Phosphorus». Inorg. Chem. 46 (10): 4028–35. doi:10.1021/ic062192q. PMID 17439206.
- ^ Robert Engel (2003-12-18). Synthesis of Carbon-Phosphorus Bonds (2 ed.). CRC Press, 2003. p. 11. ISBN 0203998243.
- ^ «Nobel Prize in Chemistry 1956 – Presentation Speech by Professor A. Ölander (committee member)». Retrieved 2009-05-05.
- ^ «Phosphorus Topics page, at Lateral Science». Archived from the original on 2009-02-21. Retrieved 2009-05-05.
- ^ a b c Emsley, John (2000). The Shocking History of Phosphorus. London: Macmillan. ISBN 0-330-39005-8.
- ^ Vanzee, Richard J.; Khan, Ahsan U. (1976). «The phosphorescence of phosphorus». The Journal of Physical Chemistry. 80 (20): 2240–2242. doi:10.1021/j100561a021.
- ^ a b Michael A. Sommers (2007-08-15). Phosphorus. The Rosen Publishing Group, 2007. p. 25. ISBN 978-1404219601.
- ^ Audi, G.; Kondev, F. G.; Wang, M.; Huang, W. J.; Naimi, S. (2017). «The NUBASE2016 evaluation of nuclear properties» (PDF). Chinese Physics C. 41 (3): 030001. Bibcode:2017ChPhC..41c0001A. doi:10.1088/1674-1137/41/3/030001.
- ^ Neufcourt, L.; Cao, Y.; Nazarewicz, W.; Olsen, E.; Viens, F. (2019). «Neutron drip line in the Ca region from Bayesian model averaging». Physical Review Letters. 122 (6): 062502–1–062502–6. arXiv:1901.07632. Bibcode:2019PhRvL.122f2502N. doi:10.1103/PhysRevLett.122.062502. PMID 30822058. S2CID 73508148.
- ^ «Phosphorus-32» (PDF). University of Michigan Department of Occupational Safety & Environmental Health. Archived from the original (PDF) on 2016-05-28. Retrieved 2010-11-18.
- ^ Koo, B.-C.; Lee, Y.-H.; Moon, D.-S.; Yoon, S.-C.; Raymond, J. C. (2013). «Phosphorus in the Young Supernova Remnant Cassiopeia A». Science. 342 (6164): 1346–8. arXiv:1312.3807. Bibcode:2013Sci…342.1346K. doi:10.1126/science.1243823. PMID 24337291. S2CID 35593706.
- ^ Rivilla, V. M.; Drozdovskaya, M. N.; Altwegg, K.; Caselli, P.; Beltrán, M. T.; Fontani, F.; van der Tak, F. F. S.; Cesaroni, R.; Vasyunin, A.; Rubin, M.; Lique, F.; Marinakis, S.; Testi, L. (2019). «ALMA and ROSINA detections of phosphorus-bearing molecules: the interstellar thread between star-forming regions and comets». Monthly Notices of the Royal Astronomical Society. 492: 1180–1198. arXiv:1911.11647. doi:10.1093/mnras/stz3336. S2CID 208290964.
- ^ ESO (15 January 2020). «Astronomers reveal interstellar thread of one of life’s building blocks». Phys.org. Retrieved 16 January 2020.
- ^ «Phosphate Rock: Statistics and Information». USGS. Retrieved 2009-06-06.
- ^ a b c d e Philpott, Tom (March–April 2013). «You Need Phosphorus to Live—and We’re Running Out». Mother Jones.
- ^ Klein, Cornelis and Cornelius S. Hurlbut, Jr., Manual of Mineralogy, Wiley, 1985, 20th ed., p. 360, ISBN 0-471-80580-7
- ^ Threlfall 1951, p. 51
- ^ Arthur D. F. Toy (2013-10-22). The Chemistry of Phosphorus. Elsevier, 2013. p. 389. ISBN 978-1483147413.
- ^ D. E. C. Corbridge «Phosphorus: An Outline of its Chemistry, Biochemistry, and Technology» 5th Edition Elsevier: Amsterdam 1995. ISBN 0-444-89307-5.
- ^ Kutzelnigg, W. (1984). «Chemical Bonding in Higher Main Group Elements» (PDF). Angew. Chem. Int. Ed. Engl. 23 (4): 272–295. doi:10.1002/anie.198402721. Archived from the original (PDF) on 2020-04-16. Retrieved 2009-05-24.
- ^ Mark, J. E.; Allcock, H. R.; West, R. «Inorganic Polymers» Prentice Hall, Englewood, NJ: 1992. ISBN 0-13-465881-7.
- ^ Heal, H. G. «The Inorganic Heterocyclic Chemistry of Sulfur, Nitrogen, and Phosphorus» Academic Press: London; 1980. ISBN 0-12-335680-6.
- ^ Weeks, Mary Elvira (1932). «The discovery of the elements. II. Elements known to the alchemists». Journal of Chemical Education. 9 (1): 11. Bibcode:1932JChEd…9…11W. doi:10.1021/ed009p11.
- ^ Beatty, Richard (2000). Phosphorus. Marshall Cavendish. p. 7. ISBN 0-7614-0946-7.
- ^ a b Schmundt, Hilmar (21 April 2010), «Experts Warn of Impending Phosphorus Crisis», Der Spiegel.
- ^ Stillman, J. M. (1960). The Story of Alchemy and Early Chemistry. New York: Dover. pp. 418–419. ISBN 0-7661-3230-7.
- ^ Baccini, Peter; Paul H. Brunner (2012-02-10). Metabolism of the Anthroposphere. MIT Press, 2012. p. 288. ISBN 978-0262300544.
- ^ Emsley, John (7 January 2002). The 13th Element: The Sordid Tale of Murder, Fire, and Phosphorus. John Wiley & Sons. ISBN 978-0-471-44149-6. Retrieved 3 February 2012.
- ^ cf. «Memoir on Combustion in General» Mémoires de l’Académie Royale des Sciences 1777, 592–600. from Henry Marshall Leicester and Herbert S. Klickstein, A Source Book in Chemistry 1400–1900 (New York: McGraw Hill, 1952)
- ^ Thomson, Robert Dundas (1870). Dictionary of chemistry with its applications to mineralogy, physiology and the arts. Rich. Griffin and Company. p. 416.
- ^ Threlfall 1951, pp. 49–66
- ^ Robert B. Heimann; Hans D. Lehmann (2015-03-10). Bioceramic Coatings for Medical Implants. John Wiley & Sons, 2015. p. 4. ISBN 978-3527684007.
- ^ The Chemistry of Phosphorus, by Arthur Toy
- ^ US patent 417943
- ^ Threlfall 1951, pp. 81–101
- ^ Parkes & Mellor 1939, p. 718–720.
- ^ a b c Threlfall 1951, pp. 167–185
- ^ Lewis R. Goldfrank; Neal Flomenbaum; Mary Ann Howland; Robert S. Hoffman; Neal A. Lewin; Lewis S. Nelson (2006). Goldfrank’s toxicologic emergencies. McGraw-Hill Professional. pp. 1486–1489. ISBN 0-07-143763-0.
- ^ The White Phosphorus Matches Prohibition Act, 1908.
- ^ «Phosphate Rock» (PDF). USGS. Retrieved 2017-03-20.
- ^ Lewis, Leo (2008-06-23). «Scientists warn of lack of vital phosphorus as biofuels raise demand». The Times.
- ^ Grantham, Jeremy (Nov 12, 2012). «Be persuasive. Be brave. Be arrested (if necessary)». Nature. 491 (7424): 303. Bibcode:2012Natur.491..303G. doi:10.1038/491303a. PMID 23151541.
- ^ a b Geeson, Michael B.; Cummins, Christopher C. (2020). «Let’s Make White Phosphorus Obsolete». ACS Central Science. 6 (6): 848–860. doi:10.1021/acscentsci.0c00332. PMC 7318074. PMID 32607432.
- ^ Tayibi, Hanan; Choura, Mohamed; López, Félix A.; Alguacil, Francisco J.; López-Delgado, Aurora (2009). «Environmental Impact and Management of Phosphogypsum». Journal of Environmental Management. 90 (8): 2377–2386. doi:10.1016/j.jenvman.2009.03.007. hdl:10261/45241. PMID 19406560.
- ^ Shriver, Atkins. Inorganic Chemistry, Fifth Edition. W. H. Freeman and Company, New York; 2010; p. 379.
- ^ «ERCO and Long Harbour». Memorial University of Newfoundland and the C.R.B. Foundation. Retrieved 2009-06-06.
- ^ Von Wagner, Rudolf (1897). Manual of chemical technology. New York: D. Appleton & Co. p. 411.
- ^ «Current EU approved additives and their E Numbers». Foods Standards Agency. 14 March 2012. Archived from the original on 21 August 2013. Retrieved 22 July 2012.
- ^ «Why is phosphoric acid used in some Coca‑Cola drinks?| Frequently Asked Questions | Coca-Cola GB». www.coca-cola.co.uk. Archived from the original on 2 August 2021. Retrieved 2021-08-31.
- ^ Moynihan, P. J. (23 November 2002). «Dietary advice in dental practice». British Dental Journal. 193 (10): 563–568. doi:10.1038/sj.bdj.4801628. PMID 12481178.
- ^ Qaseem, A; Dallas, P; Forciea, MA; Starkey, M; et al. (4 November 2014). «Dietary and pharmacologic management to prevent recurrent nephrolithiasis in adults: A clinical practice guideline from the American College of Physicians». Annals of Internal Medicine. 161 (9): 659–67. doi:10.7326/M13-2908. PMID 25364887.
- ^ a b Etesami, H. (2019). Nutrient Dynamics for Sustainable Crop Production. Springer. p. 217. ISBN 9789811386602.
- ^ «Soil Phosphorous» (PDF). United States Department of Agriculture.
- ^ «Managing Phosphorus for Crop Production». Penn State Extension. Archived from the original on 2020-10-20. Retrieved 2020-08-17.
- ^ Jessica Elzea Kogel, ed. (2006). Industrial Minerals & Rocks: Commodities, Markets, and Uses. SME, 2006. p. 964. ISBN 0873352335.
- ^ a b c d e Threlfall, R.E. (1951). 100 years of Phosphorus Making: 1851–1951. Oldbury: Albright and Wilson Ltd.
- ^ Diskowski, Herbert and Hofmann, Thomas (2005) «Phosphorus» in Ullmann’s Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim. doi:10.1002/14356007.a19_505
- ^ Roland W. Scholz; Amit H. Roy; Fridolin S. Brand; Deborah Hellums; Andrea E. Ulrich, eds. (2014-03-12). Sustainable Phosphorus Management: A Global Transdisciplinary Roadmap. Springer Science & Business Media. p. 175. ISBN 978-9400772502.
- ^ Mel Schwartz (2016-07-06). Encyclopedia and Handbook of Materials, Parts and Finishes. CRC Press. ISBN 978-1138032064.
- ^ Joseph R. Davisz, ed. (January 2001). Copper and Copper Alloys. ASM International. p. 181. ISBN 0871707268.
- ^ Hughes, J. P. W; Baron, R.; Buckland, D. H.; et al. (1962). «Phosphorus Necrosis of the Jaw: A Present-day Study: With Clinical and Biochemical Studies». Br. J. Ind. Med. 19 (2): 83–99. doi:10.1136/oem.19.2.83. PMC 1038164. PMID 14449812.
- ^ Crass, M. F. Jr. (1941). «A history of the match industry. Part 9» (PDF). Journal of Chemical Education. 18 (9): 428–431. Bibcode:1941JChEd..18..428C. doi:10.1021/ed018p428.[permanent dead link]
- ^ Oliver, Thomas (1906). «Industrial disease due to certain poisonous fumes or gases». Archives of the Public Health Laboratory. Manchester University Press. 1: 1–21.
- ^ Charnovitz, Steve (1987). «The Influence of International Labour Standards on the World Trading Regime. A Historical Overview». International Labour Review. 126 (5): 565, 571.
- ^ Alexander P. Hardt (2001). «Matches». Pyrotechnics. Post Falls Idaho USA: Pyrotechnica Publications. pp. 74–84. ISBN 0-929388-06-2.
- ^ Klaus Schrödter, Gerhard Bettermann, Thomas Staffel, Friedrich Wahl, Thomas Klein, Thomas Hofmann «Phosphoric Acid and Phosphates» in Ullmann’s Encyclopedia of Industrial Chemistry 2008, Wiley-VCH, Weinheim. doi:10.1002/14356007.a19_465.pub3
- ^ «Obsolete hand grenades». GlobalSecurity.Org. Retrieved 2009-08-03.
- ^ Dockery, Kevin (1997). Special Warfare Special Weapons. Chicago: Emperor’s Press. ISBN 1-883476-00-3.
- ^ David A. Atwood, ed. (2013-02-19). Radionuclides in the Environment. John Wiley & Sons, 2013. ISBN 978-1118632697.
- ^ Ruttenberg, K. C. Phosphorus Cycle – Terrestrial Phosphorus Cycle, Transport of Phosphorus, from Continents to the Ocean, The Marine Phosphorus Cycle. (archived link).
- ^ Lipmann, D. (1944). «Enzymatic Synthesis of Acetyl Phosphate». J Biol Chem. 155: 55–70. doi:10.1016/S0021-9258(18)43172-9.
- ^ Nelson, D. L.; Cox, M. M. «Lehninger, Principles of Biochemistry» 3rd Ed. Worth Publishing: New York, 2000. ISBN 1-57259-153-6.
- ^ Bernhardt, Nancy E.; Kasko, Artur M. (2008). Nutrition for the Middle Aged and Elderly. Nova Publishers. p. 171. ISBN 978-1-60456-146-3.
- ^ Mehanna H. M., Moledina J., Travis J. (June 2008). «Refeeding syndrome: what it is, and how to prevent and treat it». BMJ. 336 (7659): 1495–8. doi:10.1136/bmj.a301. PMC 2440847. PMID 18583681.
{{cite journal}}: CS1 maint: uses authors parameter (link) - ^ Anderson, John J. B. (1996). «Calcium, Phosphorus and Human Bone Development». Journal of Nutrition. 126 (4 Suppl): 1153S–1158S. doi:10.1093/jn/126.suppl_4.1153S. PMID 8642449.
- ^ Institute of Medicine (1997). «Phosphorus». Dietary Reference Intakes for Calcium, Phosphorus, Magnesium, Vitamin D, and Fluoride. Washington, DC: The National Academies Press. pp. 146–189. doi:10.17226/5776. ISBN 978-0-309-06403-3. PMID 23115811. S2CID 8768378.
- ^ «Overview on Dietary Reference Values for the EU population as derived by the EFSA Panel on Dietetic Products, Nutrition and Allergies» (PDF). 2017.
- ^ Tolerable Upper Intake Levels For Vitamins And Minerals (PDF), European Food Safety Authority, 2006
- ^ «Federal Register May 27, 2016 Food Labeling: Revision of the Nutrition and Supplement Facts Labels. FR page 33982» (PDF).
- ^ «Daily Value Reference of the Dietary Supplement Label Database (DSLD)». Dietary Supplement Label Database (DSLD). Archived from the original on 7 April 2020. Retrieved 16 May 2020.
- ^ Phosphorus in diet: MedlinePlus Medical Encyclopedia. Nlm.nih.gov (2011-11-07). Retrieved on 2011-11-19.
- ^ «CBRNE – Incendiary Agents, White Phosphorus (Smoking Stool Syndrome)». Retrieved 2009-05-05.
- ^ «US Navy’s Treatment of Chemical Agent Casualties and Conventional Military Chemical Injuries: FM8-285: Part 2 Conventional Military Chemical Injuries». Archived from the original on November 22, 2005. Retrieved 2009-05-05.
- ^ «CDC — NIOSH Pocket Guide to Chemical Hazards — Phosphorus (yellow)». www.cdc.gov. Retrieved 2015-11-21.
- ^ Skinner, H.F. (1990). «Methamphetamine synthesis via hydriodic acid/red phosphorus reduction of ephedrine». Forensic Science International. 48 (2): 123–134. doi:10.1016/0379-0738(90)90104-7.
- ^ a b «66 FR 52670—52675». 17 October 2001. Retrieved 2009-05-05.
- ^ «21 cfr 1309». Archived from the original on 2009-05-03. Retrieved 2009-05-05.
- ^ «21 USC, Chapter 13 (Controlled Substances Act)». Retrieved 2009-05-05.
Bibliography[edit]
- Emsley, John (2000). The Shocking history of Phosphorus. A biography of the Devil’s Element. London: MacMillan. ISBN 0-333-76638-5.
- Parkes, G. D.; Mellor, J. W. (1939). Mellor’s Modern Inorganic Chemistry. Longman’s Green and Co.
- Podger, Hugh (2002). Albright & Wilson. The Last 50 years. Studley: Brewin Books. ISBN 1-85858-223-7.
- Threlfall, Richard E. (1951). The Story of 100 years of Phosphorus Making: 1851–1951. Oldbury: Albright & Wilson Ltd.
|
Waxy white
Light red
Dark red and violet |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Phosphorus | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pronunciation | (FOS-fər-əs) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Allotropes | white, red, violet, black and others (see Allotropes of phosphorus) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Appearance | white, red and violet are waxy, black is metallic-looking | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Standard atomic weight Ar°(P) |
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Abundance | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| in the Earth’s crust | 5.2 (silicon = 100) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Phosphorus in the periodic table | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atomic number (Z) | 15 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Group | group 15 (pnictogens) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Period | period 3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Block | p-block | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electron configuration | [Ne] 3s2 3p3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electrons per shell | 2, 8, 5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Physical properties | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Phase at STP | solid | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Melting point | white: 317.3 K (44.15 °C, 111.5 °F) red: ∼860 K (∼590 °C, ∼1090 °F)[2] |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Boiling point | white: 553.7 K (280.5 °C, 536.9 °F) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Sublimation point | red: ≈689.2–863 K (≈416–590 °C, ≈780.8–1094 °F) violet: 893 K (620 °C, 1148 °F) |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density (near r.t.) | white: 1.823 g/cm3 red: ≈2.2–2.34 g/cm3 violet: 2.36 g/cm3 black: 2.69 g/cm3 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Heat of fusion | white: 0.66 kJ/mol | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Heat of vaporisation | white: 51.9 kJ/mol | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Molar heat capacity | white: 23.824 J/(mol·K) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Vapour pressure (white)
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Vapour pressure (red, b.p. 431 °C)
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atomic properties | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Oxidation states | −3, −2, −1, 0,[3] +1,[4] +2, +3, +4, +5 (a mildly acidic oxide) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electronegativity | Pauling scale: 2.19 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Ionisation energies |
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Covalent radius | 107±3 pm | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Van der Waals radius | 180 pm | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Spectral lines of phosphorus |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Other properties | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Natural occurrence | primordial | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal structure | body-centred cubic (bcc)
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Thermal conductivity | white: 0.236 W/(m⋅K) black: 12.1 W/(m⋅K) |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Magnetic ordering | white, red, violet, black: diamagnetic[5] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Molar magnetic susceptibility | −20.8×10−6 cm3/mol (293 K)[6] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Bulk modulus | white: 5 GPa red: 11 GPa |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CAS Number | 7723-14-0 (red) 12185-10-3 (white) |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| History | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Discovery | Hennig Brand (1669) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Recognised as an element by | Antoine Lavoisier[7] (1777) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Main isotopes of phosphorus
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| references |
Phosphorus is a chemical element with the symbol P and atomic number 15. Elemental phosphorus exists in two major forms, white phosphorus and red phosphorus, but because it is highly reactive, phosphorus is never found as a free element on Earth. It has a concentration in the Earth’s crust of about one gram per kilogram (compare copper at about 0.06 grams). In minerals, phosphorus generally occurs as phosphate.
Elemental phosphorus was first isolated as white phosphorus in 1669. White phosphorus emits a faint glow when exposed to oxygen – hence the name, taken from Greek mythology, Φωσφόρος meaning ‘light-bearer’ (Latin Lucifer), referring to the «Morning Star», the planet Venus. The term phosphorescence, meaning glow after illumination, derives from this property of phosphorus, although the word has since been used for a different physical process that produces a glow. The glow of phosphorus is caused by oxidation of the white (but not red) phosphorus — a process now called chemiluminescence. Together with nitrogen, arsenic, antimony, and bismuth, phosphorus is classified as a pnictogen.
Phosphorus is an element essential to sustaining life largely through phosphates, compounds containing the phosphate ion, PO43−. Phosphates are a component of DNA, RNA, ATP, and phospholipids, complex compounds fundamental to cells. Elemental phosphorus was first isolated from human urine, and bone ash was an important early phosphate source. Phosphate mines contain fossils because phosphate is present in the fossilized deposits of animal remains and excreta. Low phosphate levels are an important limit to growth in some aquatic systems. The vast majority of phosphorus compounds mined are consumed as fertilisers. Phosphate is needed to replace the phosphorus that plants remove from the soil, and its annual demand is rising nearly twice as fast as the growth of the human population. Other applications include organophosphorus compounds in detergents, pesticides, and nerve agents.
Characteristics[edit]
Allotropes[edit]
Phosphorus has several allotropes that exhibit strikingly diverse properties.[8] The two most common allotropes are white phosphorus and red phosphorus.[9]
From the perspective of applications and chemical literature, the most important form of elemental phosphorus is white phosphorus, often abbreviated as WP. It is a soft, waxy solid which consists of tetrahedral P
4 molecules, in which each atom is bound to the other three atoms by a formal single bond. This P
4 tetrahedron is also present in liquid and gaseous phosphorus up to the temperature of 800 °C (1,470 °F) when it starts decomposing to P
2 molecules.[10] The P
4 molecule in the gas phase has a P-P bond length of rg = 2.1994(3) Å as was determined by gas electron diffraction.[11] The nature of bonding in this P
4 tetrahedron can be described by spherical aromaticity or cluster bonding, that is the electrons are highly delocalized. This has been illustrated by calculations of the magnetically induced currents, which sum up to 29 nA/T, much more than in the archetypical aromatic molecule benzene (11 nA/T).[11]
White phosphorus exists in two crystalline forms: α (alpha) and β (beta). At room temperature, the α-form is stable. It is more common, has cubic crystal structure and at 195.2 K (−78.0 °C), it transforms into β-form, which has hexagonal crystal structure. These forms differ in terms of the relative orientations of the constituent P4 tetrahedra.[12][13] The β form of white phosphorus contains three slightly different P
4 molecules, i.e. 18 different P-P bond lengths between 2.1768(5) and 2.1920(5) Å. The average P-P bond length is 2.183(5) Å.[14]
White phosphorus is the least stable, the most reactive, the most volatile, the least dense and the most toxic of the allotropes. White phosphorus gradually changes to red phosphorus. This transformation is accelerated by light and heat, and samples of white phosphorus almost always contain some red phosphorus and accordingly appear yellow. For this reason, white phosphorus that is aged or otherwise impure (e.g., weapons-grade, not lab-grade WP) is also called yellow phosphorus. When exposed to oxygen, white phosphorus glows in the dark with a very faint tinge of green and blue. It is highly flammable and pyrophoric (self-igniting) upon contact with air. Owing to its pyrophoricity, white phosphorus is used as an additive in napalm. The odour of combustion of this form has a characteristic garlic smell, and samples are commonly coated with white phosphorus pentoxide, which consists of P
4O
10 tetrahedra with oxygen inserted between the phosphorus atoms and at their vertices. White phosphorus is insoluble in water but soluble in carbon disulfide.[15]
Thermal decomposition of P4 at 1100 K gives diphosphorus, P2. This species is not stable as a solid or liquid. The dimeric unit contains a triple bond and is analogous to N2. It can also be generated as a transient intermediate in solution by thermolysis of organophosphorus precursor reagents.[16] At still higher temperatures, P2 dissociates into atomic P.[15]
| Form | white(α) | white(β) | red | violet | black |
|---|---|---|---|---|---|
| Symmetry | Body-centred cubic |
Triclinic | Amorphous | Monoclinic | Orthorhombic |
| Pearson symbol | aP24 | mP84 | oS8 | ||
| Space group | I43m | P1 No.2 | P2/c No.13 | Cmca No.64 | |
| Density (g/cm3) | 1.828 | 1.88 | ~2.2 | 2.36 | 2.69 |
| Band gap (eV) | 2.1 | 1.8 | 1.5 | 0.34 | |
| Refractive index | 1.8244 | 2.6 | 2.4 |
Red phosphorus is polymeric in structure. It can be viewed as a derivative of P4 wherein one P-P bond is broken, and one additional bond is formed with the neighbouring tetrahedron resulting in chains of P21 molecules linked by van der Waals forces.[18] Red phosphorus may be formed by heating white phosphorus to 250 °C (482 °F) or by exposing white phosphorus to sunlight.[19] Phosphorus after this treatment is amorphous. Upon further heating, this material crystallises. In this sense, red phosphorus is not an allotrope, but rather an intermediate phase between the white and violet phosphorus, and most of its properties have a range of values. For example, freshly prepared, bright red phosphorus is highly reactive and ignites at about 300 °C (572 °F),[20] though it is more stable than white phosphorus, which ignites at about 30 °C (86 °F).[21] After prolonged heating or storage, the color darkens (see infobox images); the resulting product is more stable and does not spontaneously ignite in air.[22]
Violet phosphorus is a form of phosphorus that can be produced by day-long annealing of red phosphorus above 550 °C. In 1865, Hittorf discovered that when phosphorus was recrystallised from molten lead, a red/purple form is obtained. Therefore, this form is sometimes known as «Hittorf’s phosphorus» (or violet or α-metallic phosphorus).[17]
Black phosphorus is the least reactive allotrope and the thermodynamically stable form below 550 °C (1,022 °F). It is also known as β-metallic phosphorus and has a structure somewhat resembling that of graphite.[23][24] It is obtained by heating white phosphorus under high pressures (about 12,000 standard atmospheres or 1.2 gigapascals). It can also be produced at ambient conditions using metal salts, e.g. mercury, as catalysts.[25] In appearance, properties, and structure, it resembles graphite, being black and flaky, a conductor of electricity, and has puckered sheets of linked atoms.[26]
Another form, scarlet phosphorus, is obtained by allowing a solution of white phosphorus in carbon disulfide to evaporate in sunlight.[17]
Chemiluminescence[edit]
White phosphorus exposed to air glows in the dark
When first isolated, it was observed that the green glow emanating from white phosphorus would persist for a time in a stoppered jar, but then cease. Robert Boyle in the 1680s ascribed it to «debilitation» of the air. Actually, it is oxygen being consumed. By the 18th century, it was known that in pure oxygen, phosphorus does not glow at all;[27] there is only a range of partial pressures at which it does. Heat can be applied to drive the reaction at higher pressures.[28]
In 1974, the glow was explained by R. J. van Zee and A. U. Khan.[29][30] A reaction with oxygen takes place at the surface of the solid (or liquid) phosphorus, forming the short-lived molecules HPO and P
2O
2 that both emit visible light. The reaction is slow and only very little of the intermediates are required to produce the luminescence, hence the extended time the glow continues in a stoppered jar.
Since its discovery, phosphors and phosphorescence were used loosely to describe substances that shine in the dark without burning. Although the term phosphorescence is derived from phosphorus, the reaction that gives phosphorus its glow is properly called chemiluminescence (glowing due to a cold chemical reaction), not phosphorescence (re-emitting light that previously fell onto a substance and excited it).[31]
Isotopes[edit]
There are 23 known isotopes of phosphorus,[32] ranging from 25
P to 47
P.[33] Only 31
P is stable and is therefore present at 100% abundance. The half-integer nuclear spin and high abundance of 31P make phosphorus-31 NMR spectroscopy a very useful analytical tool in studies of phosphorus-containing samples.
Two radioactive isotopes of phosphorus have half-lives suitable for biological scientific experiments. These are:
- 32
P, a beta-emitter (1.71 MeV) with a half-life of 14.3 days, which is used routinely in life-science laboratories, primarily to produce radiolabeled DNA and RNA probes, e.g. for use in Northern blots or Southern blots. - 33
P, a beta-emitter (0.25 MeV) with a half-life of 25.4 days. It is used in life-science laboratories in applications in which lower energy beta emissions are advantageous such as DNA sequencing.
The high energy beta particles from 32
P penetrate skin and corneas and any 32
P ingested, inhaled, or absorbed is readily incorporated into bone and nucleic acids. For these reasons, Occupational Safety and Health Administration in the United States, and similar institutions in other developed countries require personnel working with 32
P to wear lab coats, disposable gloves, and safety glasses or goggles to protect the eyes, and avoid working directly over open containers. Monitoring personal, clothing, and surface contamination is also required. Shielding requires special consideration. The high energy of the beta particles gives rise to secondary emission of X-rays via Bremsstrahlung (braking radiation) in dense shielding materials such as lead. Therefore, the radiation must be shielded with low density materials such as acrylic or other plastic, water, or (when transparency is not required), even wood.[34]
Occurrence[edit]
Universe[edit]
In 2013, astronomers detected phosphorus in Cassiopeia A, which confirmed that this element is produced in supernovae as a byproduct of supernova nucleosynthesis. The phosphorus-to-iron ratio in material from the supernova remnant could be up to 100 times higher than in the Milky Way in general.[35]
In 2020, astronomers analysed ALMA and ROSINA data from the massive star-forming region AFGL 5142, to detect phosphorus-bearing molecules and how they are carried in comets to the early Earth.[36][37]
Crust and organic sources[edit]
Phosphorus has a concentration in the Earth’s crust of about one gram per kilogram (compare copper at about 0.06 grams). It is not found free in nature, but is widely distributed in many minerals, usually as phosphates.[9] Inorganic phosphate rock, which is partially made of apatite (a group of minerals being, generally, pentacalcium triorthophosphate fluoride (hydroxide)), is today the chief commercial source of this element. According to the US Geological Survey (USGS), about 50 percent of the global phosphorus reserves are in the Arab nations.[38] 85% of Earth’s known reserves are in Morocco with smaller deposits in China, Russia,[39] Florida, Idaho, Tennessee, Utah, and elsewhere.[40] Albright and Wilson in the UK and their Niagara Falls plant, for instance, were using phosphate rock in the 1890s and 1900s from Tennessee, Florida, and the Îles du Connétable (guano island sources of phosphate); by 1950, they were using phosphate rock mainly from Tennessee and North Africa.[41]
Organic sources, namely urine, bone ash and (in the latter 19th century) guano, were historically of importance but had only limited commercial success.[42] As urine contains phosphorus, it has fertilising qualities which are still harnessed today in some countries, including Sweden, using methods for reuse of excreta. To this end, urine can be used as a fertiliser in its pure form or part of being mixed with water in the form of sewage or sewage sludge.
Compounds[edit]
Phosphorus(V)[edit]
The tetrahedral structure of P4O10 and P4S10.
The most prevalent compounds of phosphorus are derivatives of phosphate (PO43−), a tetrahedral anion.[43] Phosphate is the conjugate base of phosphoric acid, which is produced on a massive scale for use in fertilisers. Being triprotic, phosphoric acid converts stepwise to three conjugate bases:
- H3PO4 + H2O ⇌ H3O+ + H2PO4− Ka1 = 7.25×10−3
- H2PO4− + H2O ⇌ H3O+ + HPO42− Ka2 = 6.31×10−8
- HPO42− + H2O ⇌ H3O+ + PO43− Ka3 = 3.98×10−13
Phosphate exhibits a tendency to form chains and rings containing P-O-P bonds. Many polyphosphates are known, including ATP. Polyphosphates arise by dehydration of hydrogen phosphates such as HPO42− and H2PO4−. For example, the industrially important pentasodium triphosphate (also known as sodium tripolyphosphate, STPP) is produced industrially on by the megatonne by this condensation reaction:
- 2 Na2[(HO)PO3] + Na[(HO)2PO2] → Na5[O3P-O-P(O)2-O-PO3] + 2 H2O
Phosphorus pentoxide (P4O10) is the acid anhydride of phosphoric acid, but several intermediates between the two are known. This waxy white solid reacts vigorously with water.
With metal cations, phosphate forms a variety of salts. These solids are polymeric, featuring P-O-M linkages. When the metal cation has a charge of 2+ or 3+, the salts are generally insoluble, hence they exist as common minerals. Many phosphate salts are derived from hydrogen phosphate (HPO42−).
PCl5 and PF5 are common compounds. PF5 is a colourless gas and the molecules have trigonal bipyramidal geometry. PCl5 is a colourless solid which has an ionic formulation of PCl4+ PCl6−, but adopts the trigonal bipyramidal geometry when molten or in the vapour phase.[15] PBr5 is an unstable solid formulated as PBr4+Br−and PI5 is not known.[15] The pentachloride and pentafluoride are Lewis acids. With fluoride, PF5 forms PF6−, an anion that is isoelectronic with SF6. The most important oxyhalide is phosphorus oxychloride, (POCl3), which is approximately tetrahedral.
Before extensive computer calculations were feasible, it was thought that bonding in phosphorus(V) compounds involved d orbitals. Computer modeling of molecular orbital theory indicates that this bonding involves only s- and p-orbitals.[44]
Phosphorus(III)[edit]
All four symmetrical trihalides are well known: gaseous PF3, the yellowish liquids PCl3 and PBr3, and the solid PI3. These materials are moisture sensitive, hydrolysing to give phosphorous acid. The trichloride, a common reagent, is produced by chlorination of white phosphorus:
- P4 + 6 Cl2 → 4 PCl3
The trifluoride is produced from the trichloride by halide exchange. PF3 is toxic because it binds to haemoglobin.
Phosphorus(III) oxide, P4O6 (also called tetraphosphorus hexoxide) is the anhydride of P(OH)3, the minor tautomer of phosphorous acid. The structure of P4O6 is like that of P4O10 without the terminal oxide groups.
Phosphorus(I) and phosphorus(II)[edit]
These compounds generally feature P–P bonds.[15] Examples include catenated derivatives of phosphine and organophosphines. Compounds containing P=P double bonds have also been observed, although they are rare.
Phosphides and phosphines[edit]
Phosphides arise by reaction of metals with red phosphorus. The alkali metals (group 1) and alkaline earth metals can form ionic compounds containing the phosphide ion, P3−. These compounds react with water to form phosphine. Other phosphides, for example Na3P7, are known for these reactive metals. With the transition metals as well as the monophosphides there are metal-rich phosphides, which are generally hard refractory compounds with a metallic lustre, and phosphorus-rich phosphides which are less stable and include semiconductors.[15] Schreibersite is a naturally occurring metal-rich phosphide found in meteorites. The structures of the metal-rich and phosphorus-rich phosphides can be complex.
Phosphine (PH3) and its organic derivatives (PR3) are structural analogues of ammonia (NH3), but the bond angles at phosphorus are closer to 90° for phosphine and its organic derivatives. It is an ill-smelling, toxic compound. Phosphorus has an oxidation number of −3 in phosphine. Phosphine is produced by hydrolysis of calcium phosphide, Ca3P2. Unlike ammonia, phosphine is oxidised by air. Phosphine is also far less basic than ammonia. Other phosphines are known which contain chains of up to nine phosphorus atoms and have the formula PnHn+2.[15] The highly flammable gas diphosphine (P2H4) is an analogue of hydrazine.
Oxoacids[edit]
Phosphorous oxoacids are extensive, often commercially important, and sometimes structurally complicated. They all have acidic protons bound to oxygen atoms, some have nonacidic protons that are bonded directly to phosphorus and some contain phosphorus — phosphorus bonds.[15] Although many oxoacids of phosphorus are formed, only nine are commercially important, and three of them, hypophosphorous acid, phosphorous acid, and phosphoric acid, are particularly important.
| Oxidation state | Formula | Name | Acidic protons | Compounds |
|---|---|---|---|---|
| +1 | HH2PO2 | hypophosphorous acid | 1 | acid, salts |
| +3 | H2HPO3 | phosphorous acid | 2 | acid, salts |
| +3 | HPO2 | metaphosphorous acid | 1 | salts |
| +3 | H3PO3 | (ortho)phosphorous acid | 3 | acid, salts |
| +4 | H4P2O6 | hypophosphoric acid | 4 | acid, salts |
| +5 | (HPO3)n | metaphosphoric acids | n | salts (n = 3,4,6) |
| +5 | H(HPO3)nOH | polyphosphoric acids | n+2 | acids, salts (n = 1-6) |
| +5 | H5P3O10 | tripolyphosphoric acid | 3 | salts |
| +5 | H4P2O7 | pyrophosphoric acid | 4 | acid, salts |
| +5 | H3PO4 | (ortho)phosphoric acid | 3 | acid, salts |
Nitrides[edit]
The PN molecule is considered unstable, but is a product of crystalline phosphorus nitride decomposition at 1100 K. Similarly, H2PN is considered unstable, and phosphorus nitride halogens like F2PN, Cl2PN, Br2PN, and I2PN oligomerise into cyclic Polyphosphazenes. For example, compounds of the formula (PNCl2)n exist mainly as rings such as the trimer hexachlorophosphazene. The phosphazenes arise by treatment of phosphorus pentachloride with ammonium chloride:
PCl5 + NH4Cl → 1/n (NPCl2)n + 4 HCl
When the chloride groups are replaced by alkoxide (RO−), a family of polymers is produced with potentially useful properties.[45]
Sulfides[edit]
Phosphorus forms a wide range of sulfides, where the phosphorus can be in P(V), P(III) or other oxidation states. The three-fold symmetric P4S3 is used in strike-anywhere matches. P4S10 and P4O10 have analogous structures.[46] Mixed oxyhalides and oxyhydrides of phosphorus(III) are almost unknown.
Organophosphorus compounds[edit]
Compounds with P-C and P-O-C bonds are often classified as organophosphorus compounds. They are widely used commercially. The PCl3 serves as a source of P3+ in routes to organophosphorus(III) compounds. For example, it is the precursor to triphenylphosphine:
- PCl3 + 6 Na + 3 C6H5Cl → P(C6H5)3 + 6 NaCl
Treatment of phosphorus trihalides with alcohols and phenols gives phosphites, e.g. triphenylphosphite:
- PCl3 + 3 C6H5OH → P(OC6H5)3 + 3 HCl
Similar reactions occur for phosphorus oxychloride, affording triphenylphosphate:
- OPCl3 + 3 C6H5OH → OP(OC6H5)3 + 3 HCl
History[edit]
Etymology[edit]
The name Phosphorus in Ancient Greece was the name for the planet Venus and is derived from the Greek words (φῶς = light, φέρω = carry), which roughly translates as light-bringer or light carrier.[19] (In Greek mythology and tradition, Augerinus (Αυγερινός = morning star, still in use today), Hesperus or Hesperinus (΄Εσπερος or Εσπερινός or Αποσπερίτης = evening star, still in use today) and Eosphorus (Εωσφόρος = dawnbearer, not in use for the planet after Christianity) are close homologues, and also associated with Phosphorus-the-morning-star).
According to the Oxford English Dictionary, the correct spelling of the element is phosphorus. The word phosphorous is the adjectival form of the P3+ valence: so, just as sulfur forms sulfurous and sulfuric compounds, phosphorus forms phosphorous compounds (e.g., phosphorous acid) and P5+ valence phosphoric compounds (e.g., phosphoric acids and phosphates).
Discovery[edit]
The discovery of phosphorus, the first element to be discovered that was not known since ancient times,[47] is credited to the German alchemist Hennig Brand in 1669, although others might have discovered phosphorus around the same time.[48] Brand experimented with urine, which contains considerable quantities of dissolved phosphates from normal metabolism.[19] Working in Hamburg, Brand attempted to create the fabled philosopher’s stone through the distillation of some salts by evaporating urine, and in the process produced a white material that glowed in the dark and burned brilliantly. It was named phosphorus mirabilis («miraculous bearer of light»).[49]
Brand’s process originally involved letting urine stand for days until it gave off a terrible smell. Then he boiled it down to a paste, heated this paste to a high temperature, and led the vapours through water, where he hoped they would condense to gold. Instead, he obtained a white, waxy substance that glowed in the dark. Brand had discovered phosphorus. Specifically, Brand produced ammonium sodium hydrogen phosphate, (NH
4)NaHPO
4. While the quantities were essentially correct (it took about 1,100 litres [290 US gal] of urine to make about 60 g of phosphorus), it was unnecessary to allow the urine to rot first. Later scientists discovered that fresh urine yielded the same amount of phosphorus.[31]
Brand at first tried to keep the method secret,[50] but later sold the recipe for 200 thalers to D. Krafft from Dresden.[19] Krafft toured much of Europe with it, including England, where he met with Robert Boyle. The secret—that the substance was made from urine—leaked out, and Johann Kunckel (1630–1703) was able to reproduce it in Sweden (1678). Later, Boyle in London (1680) also managed to make phosphorus, possibly with the aid of his assistant, Ambrose Godfrey-Hanckwitz. Godfrey later made a business of the manufacture of phosphorus.
Boyle states that Krafft gave him no information as to the preparation of phosphorus other than that it was derived from «somewhat that belonged to the body of man». This gave Boyle a valuable clue, so that he, too, managed to make phosphorus, and published the method of its manufacture.[19] Later he improved Brand’s process by using sand in the reaction (still using urine as base material),
- 4 NaPO
3 + 2 SiO
2 + 10 C → 2 Na
2SiO
3 + 10 CO + P
4
Robert Boyle was the first to use phosphorus to ignite sulfur-tipped wooden splints, forerunners of our modern matches, in 1680.[51]
Phosphorus was the 13th element to be discovered. Because of its tendency to spontaneously combust when left alone in air, it is sometimes referred to as «the Devil’s element».[52]
Bone ash and guano[edit]
Antoine Lavoisier recognized phosphorus as an element in 1777 after Johan Gottlieb Gahn and Carl Wilhelm Scheele, in 1769, showed that calcium phosphate (Ca
3(PO
4)
2) is found in bones by obtaining elemental phosphorus from bone ash.[53]
Bone ash was the major source of phosphorus until the 1840s. The method started by roasting bones, then employed the use of fire clay retorts encased in a very hot brick furnace to distill out the highly toxic elemental phosphorus product.[54] Alternately, precipitated phosphates could be made from ground-up bones that had been de-greased and treated with strong acids. White phosphorus could then be made by heating the precipitated phosphates, mixed with ground coal or charcoal in an iron pot, and distilling off phosphorus vapour in a retort.[55] Carbon monoxide and other flammable gases produced during the reduction process were burnt off in a flare stack.
In the 1840s, world phosphate production turned to the mining of tropical island deposits formed from bird and bat guano (see also Guano Islands Act). These became an important source of phosphates for fertiliser in the latter half of the 19th century.[56]
Phosphate rock[edit]
Phosphate rock, which usually contains calcium phosphate, was first used in 1850 to make phosphorus, and following the introduction of the electric arc furnace by James Burgess Readman in 1888[57] (patented 1889),[58] elemental phosphorus production switched from the bone-ash heating, to electric arc production from phosphate rock. After the depletion of world guano sources about the same time, mineral phosphates became the major source of phosphate fertiliser production. Phosphate rock production greatly increased after World War II, and remains the primary global source of phosphorus and phosphorus chemicals today. See the article on peak phosphorus for more information on the history and present state of phosphate mining. Phosphate rock remains a feedstock in the fertiliser industry, where it is treated with sulfuric acid to produce various «superphosphate» fertiliser products.
Incendiaries[edit]
White phosphorus was first made commercially in the 19th century for the match industry. This used bone ash for a phosphate source, as described above. The bone-ash process became obsolete when the submerged-arc furnace for phosphorus production was introduced to reduce phosphate rock.[59][60] The electric furnace method allowed production to increase to the point where phosphorus could be used in weapons of war.[29][61] In World War I, it was used in incendiaries, smoke screens and tracer bullets.[61] A special incendiary bullet was developed to shoot at hydrogen-filled Zeppelins over Britain (hydrogen being highly flammable).[61] During World War II, Molotov cocktails made of phosphorus dissolved in petrol were distributed in Britain to specially selected civilians within the British resistance operation, for defence; and phosphorus incendiary bombs were used in war on a large scale. Burning phosphorus is difficult to extinguish and if it splashes onto human skin it has horrific effects.[15]
Early matches used white phosphorus in their composition, which was dangerous due to its toxicity. Murders, suicides and accidental poisonings resulted from its use. (An apocryphal tale tells of a woman attempting to murder her husband with white phosphorus in his food, which was detected by the stew’s giving off luminous steam).[29] In addition, exposure to the vapours gave match workers a severe necrosis of the bones of the jaw, known as «phossy jaw». When a safe process for manufacturing red phosphorus was discovered, with its far lower flammability and toxicity, laws were enacted, under the Berne Convention (1906), requiring its adoption as a safer alternative for match manufacture.[62] The toxicity of white phosphorus led to discontinuation of its use in matches.[63] The Allies used phosphorus incendiary bombs in World War II to destroy Hamburg, the place where the «miraculous bearer of light» was first discovered.[49]
Production[edit]
Mining of phosphate rock in Nauru
In 2017, the USGS estimated 68 billion tons of world reserves, where reserve figures refer to the amount assumed recoverable at current market prices; 0.261 billion tons were mined in 2016.[64] Critical to contemporary agriculture, its annual demand is rising nearly twice as fast as the growth of the human population.[39] The production of phosphorus may have peaked before 2011 and some scientists predict reserves will be depleted in before the end of the 21st Century.»[65][39][66] Phosphorus comprises about 0.1% by mass of the average rock, and consequently, the Earth’s supply is vast, though dilute.[15]
Wet process[edit]
Most phosphorus-bearing material is for agriculture fertilisers. In this case where the standards of purity are modest, phosphorus is obtained from phosphate rock by what is called the «wet process.» The minerals are treated with sulfuric acid to give phosphoric acid. Phosphoric acid is then neutralized to give various phosphate salts, which comprise fertilizers. In the wet process, phosphorus does not undergo redox.[67] About five tons of phosphogypsum waste are generated per ton of phosphoric acid production. Annually, the estimated generation of phosphogypsum worldwide is 100 to 280 Mt.[68]
Thermal process[edit]
For the use of phosphorus in drugs, detergents, and foodstuff, the standards of purity are high, which led to the development of the thermal process. In this process, phosphate minerals are converted to white phosphorus, which can be purified by distillation. The white phosphorus is then oxidised to phosphoric acid and subsequently neutralised with a base to give phosphate salts. The thermal process is energy intensive.[67] Presently, about 1,000,000 short tons (910,000 t) of elemental phosphorus is produced annually. Calcium phosphate (phosphate rock), mostly mined in Florida and North Africa, can be heated to 1,200–1,500 °C with sand, which is mostly SiO
2, and coke to produce P
4. The P
4 product, being volatile, is readily isolated:[69]
- 4 Ca5(PO4)3F + 18 SiO2 + 30 C → 3 P4 + 30 CO + 18 CaSiO3 + 2 CaF2
- 2 Ca3(PO4)2 + 6 SiO2 + 10 C → 6 CaSiO3 + 10 CO + P4
Side products from the thermal process include ferrophosphorus, a crude form of Fe2P, resulting from iron impurities in the mineral precursors. The silicate slag is a useful construction material. The fluoride is sometimes recovered for use in water fluoridation. More problematic is a «mud» containing significant amounts of white phosphorus. Production of white phosphorus is conducted in large facilities in part because it is energy intensive. The white phosphorus is transported in molten form. Some major accidents have occurred during transportation.[70]
Historical routes[edit]
Historically, before the development of mineral-based extractions, white phosphorus was isolated on an industrial scale from bone ash.[71] In this process, the tricalcium phosphate in bone ash is converted to monocalcium phosphate with sulfuric acid:
- Ca3(PO4)2 + 2 H2SO4 → Ca(H2PO4)2 + 2 CaSO4
Monocalcium phosphate is then dehydrated to the corresponding metaphosphate:
- Ca(H2PO4)2 → Ca(PO3)2 + 2 H2O
When ignited to a white heat (~1300C) with charcoal, calcium metaphosphate yields two-thirds of its weight of white phosphorus while one-third of the phosphorus remains in the residue as calcium orthophosphate:
- 3 Ca(PO3)2 + 10 C → Ca3(PO4)2 + 10 CO + P4
Applications[edit]
Food additive[edit]
Phosphorus is an essential mineral for humans listed in the Dietary Reference Intake (DRI).
Food-grade phosphoric acid (additive E338[72]) is used to acidify foods and beverages such as various colas and jams, providing a tangy or sour taste. The phosphoric acid also serves as a preservative.[73] Soft drinks containing phosphoric acid, which would include Coca-Cola, are sometimes called phosphate sodas or phosphates. Phosphoric acid in soft drinks has the potential to cause dental erosion.[74] Phosphoric acid also has the potential to contribute to the formation of kidney stones, especially in those who have had kidney stones previously.[75]
Fertiliser[edit]
Phosphorus is an essential plant nutrient (the most often limiting nutrient, after nitrogen),[76] and the bulk of all phosphorus production is in concentrated phosphoric acids for agriculture fertilisers, containing as much as 70% to 75% P2O5. That led to large increase in phosphate (PO43−) production in the second half of the 20th century.[39] Artificial phosphate fertilisation is necessary because phosphorus is essential to all living organisms; it is involved in energy transfers, strength of root and stems, photosynthesis, the expansion of plant roots, formation of seeds and flowers, and other important factors effecting overall plant health and genetics.[76]
Natural phosphorus-bearing compounds are mostly inaccessible to plants because of the low solubility and mobility in soil.[77] Most phosphorus is very stable in the soil minerals or organic matter of the soil. Even when phosphorus is added in manure or fertilizer it can become fixed in the soil. Therefore, the natural cycle of phosphorus is very slow. Some of the fixed phosphorus is released again over time, sustaining wild plant growth, however, more is needed to sustain intensive cultivation of crops.[78] Fertiliser is often in the form of superphosphate of lime, a mixture of calcium dihydrogen phosphate (Ca(H2PO4)2), and calcium sulfate dihydrate (CaSO4·2H2O) produced reacting sulfuric acid and water with calcium phosphate.
Processing phosphate minerals with sulfuric acid for obtaining fertiliser is so important to the global economy that this is the primary industrial market for sulfuric acid and the greatest industrial use of elemental sulfur.[79]
| Widely used compounds | Use |
|---|---|
| Ca(H2PO4)2·H2O | Baking powder and fertilisers |
| CaHPO4·2H2O | Animal food additive, toothpowder |
| H3PO4 | Manufacture of phosphate fertilisers |
| PCl3 | Manufacture of POCl3 and pesticides |
| POCl3 | Manufacture of plasticiser |
| P4S10 | Manufacturing of additives and pesticides |
| Na5P3O10 | Detergents |
Organophosphorus[edit]
White phosphorus is widely used to make organophosphorus compounds through intermediate phosphorus chlorides and two phosphorus sulfides, phosphorus pentasulfide and phosphorus sesquisulfide.[80] Organophosphorus compounds have many applications, including in plasticisers, flame retardants, pesticides, extraction agents, nerve agents and water treatment.[15][81]
Metallurgical aspects[edit]
Phosphorus is also an important component in steel production, in the making of phosphor bronze, and in many other related products.[82][83] Phosphorus is added to metallic copper during its smelting process to react with oxygen present as an impurity in copper and to produce phosphorus-containing copper (CuOFP) alloys with a higher hydrogen embrittlement resistance than normal copper.[84]
Matches[edit]
Match striking surface made of a mixture of red phosphorus, glue and ground glass. The glass powder is used to increase the friction.
The first striking match with a phosphorus head was invented by Charles Sauria in 1830. These matches (and subsequent modifications) were made with heads of white phosphorus, an oxygen-releasing compound (potassium chlorate, lead dioxide, or sometimes nitrate), and a binder. They were poisonous to the workers in manufacture,[85] sensitive to storage conditions, toxic if ingested, and hazardous when accidentally ignited on a rough surface.[86][87] Production in several countries was banned between 1872 and 1925.[88] The international Berne Convention, ratified in 1906, prohibited the use of white phosphorus in matches.
In consequence, phosphorous matches were gradually replaced by safer alternatives. Around 1900 French chemists Henri Sévène and Emile David Cahen invented the modern strike-anywhere match, wherein the white phosphorus was replaced by phosphorus sesquisulfide (P4S3), a non-toxic and non-pyrophoric compound that ignites under friction. For a time these safer strike-anywhere matches were quite popular but in the long run they were superseded by the modern safety match.
Safety matches are very difficult to ignite on any surface other than a special striker strip. The strip contains non-toxic red phosphorus and the match head potassium chlorate, an oxygen-releasing compound. When struck, small amounts of abrasion from match head and striker strip are mixed intimately to make a small quantity of Armstrong’s mixture, a very touch sensitive composition. The fine powder ignites immediately and provides the initial spark to set off the match head. Safety matches separate the two components of the ignition mixture until the match is struck. This is the key safety advantage as it prevents accidental ignition. Nonetheless, safety matches, invented in 1844 by Gustaf Erik Pasch and market ready by the 1860s, didn’t gain consumer acceptance until the prohibition of white phosphorus. Using a dedicated striker strip was considered clumsy.[20][80][89]
Water softening[edit]
Sodium tripolyphosphate made from phosphoric acid is used in laundry detergents in some countries, but banned for this use in others.[22] This compound softens the water to enhance the performance of the detergents and to prevent pipe/boiler tube corrosion.[90]
Miscellaneous[edit]
- Phosphates are used to make special glasses for sodium lamps.[22]
- Bone-ash, calcium phosphate, is used in the production of fine china.[22]
- Phosphoric acid made from elemental phosphorus is used in food applications such as soft drinks, and as a starting point for food grade phosphates.[80] These include mono-calcium phosphate for baking powder and sodium tripolyphosphate.[80] Phosphates are used to improve the characteristics of processed meat and cheese, and in toothpaste.[80]
- White phosphorus, called «WP» (slang term «Willie Peter») is used in military applications as incendiary bombs, for smoke-screening as smoke pots and smoke bombs, and in tracer ammunition. It is also a part of an obsolete M34 White Phosphorus US hand grenade. This multipurpose grenade was mostly used for signaling, smoke screens, and inflammation; it could also cause severe burns and had a psychological impact on the enemy.[91][92] Military uses of white phosphorus are constrained by international law.
- 32P and 33P are used as radioactive tracers in biochemical laboratories.[93]
Biological role[edit]
Inorganic phosphorus in the form of the phosphate PO3−
4 is required for all known forms of life.[94] Phosphorus plays a major role in the structural framework of DNA and RNA. Living cells use phosphate to transport cellular energy with adenosine triphosphate (ATP), necessary for every cellular process that uses energy. ATP is also important for phosphorylation, a key regulatory event in cells. Phospholipids are the main structural components of all cellular membranes. Calcium phosphate salts assist in stiffening bones.[15] Biochemists commonly use the abbreviation «Pi» to refer to inorganic phosphate.[95]
Every living cell is encased in a membrane that separates it from its surroundings. Cellular membranes are composed of a phospholipid matrix and proteins, typically in the form of a bilayer. Phospholipids are derived from glycerol with two of the glycerol hydroxyl (OH) protons replaced by fatty acids as an ester, and the third hydroxyl proton has been replaced with phosphate bonded to another alcohol.[96]
An average adult human contains about 0.7 kg of phosphorus, about 85–90% in bones and teeth in the form of apatite, and the remainder in soft tissues and extracellular fluids (~1%). The phosphorus content increases from about 0.5% by mass in infancy to 0.65–1.1% by mass in adults. Average phosphorus concentration in the blood is about 0.4 g/L, about 70% of that is organic and 30% inorganic phosphates.[97] An adult with healthy diet consumes and excretes about 1–3 grams of phosphorus per day, with consumption in the form of inorganic phosphate and phosphorus-containing biomolecules such as nucleic acids and phospholipids; and excretion almost exclusively in the form of phosphate ions such as H
2PO−
4 and HPO2−
4. Only about 0.1% of body phosphate circulates in the blood, paralleling the amount of phosphate available to soft tissue cells.
Bone and teeth enamel[edit]
The main component of bone is hydroxyapatite as well as amorphous forms of calcium phosphate, possibly including carbonate. Hydroxyapatite is the main component of tooth enamel. Water fluoridation enhances the resistance of teeth to decay by the partial conversion of this mineral to the still harder material called fluoroapatite:[15]
- Ca
5(PO
4)
3OH + F−
→ Ca
5(PO
4)
3F + OH−
Phosphorus deficiency[edit]
In medicine, phosphate deficiency syndrome may be caused by malnutrition, by failure to absorb phosphate, and by metabolic syndromes that draw phosphate from the blood (such as in refeeding syndrome after malnutrition[98]) or passing too much of it into the urine. All are characterised by hypophosphatemia, which is a condition of low levels of soluble phosphate levels in the blood serum and inside the cells. Symptoms of hypophosphatemia include neurological dysfunction and disruption of muscle and blood cells due to lack of ATP. Too much phosphate can lead to diarrhoea and calcification (hardening) of organs and soft tissue, and can interfere with the body’s ability to use iron, calcium, magnesium, and zinc.[99]
Phosphorus is an essential macromineral for plants, which is studied extensively in edaphology to understand plant uptake from soil systems. Phosphorus is a limiting factor in many ecosystems; that is, the scarcity of phosphorus limits the rate of organism growth. An excess of phosphorus can also be problematic, especially in aquatic systems where eutrophication sometimes leads to algal blooms.[39]
Nutrition[edit]
Dietary recommendations[edit]
The U.S. Institute of Medicine (IOM) updated Estimated Average Requirements (EARs) and Recommended Dietary Allowances (RDAs) for phosphorus in 1997. If there is not sufficient information to establish EARs and RDAs, an estimate designated Adequate Intake (AI) is used instead. The current EAR for phosphorus for people ages 19 and up is 580 mg/day. The RDA is 700 mg/day. RDAs are higher than EARs so as to identify amounts that will cover people with higher than average requirements. RDA for pregnancy and lactation are also 700 mg/day. For people ages 1–18 years the RDA increases with age from 460 to 1250 mg/day. As for safety, the IOM sets Tolerable upper intake levels (ULs) for vitamins and minerals when evidence is sufficient. In the case of phosphorus the UL is 4000 mg/day. Collectively the EARs, RDAs, AIs and ULs are referred to as Dietary Reference Intakes (DRIs).[100]
The European Food Safety Authority (EFSA) refers to the collective set of information as Dietary Reference Values, with Population Reference Intake (PRI) instead of RDA, and Average Requirement instead of EAR. AI and UL defined the same as in United States. For people ages 15 and older, including pregnancy and lactation, the AI is set at 550 mg/day. For children ages 4–10 the AI is 440 mg/day, and for ages 11–17 it is 640 mg/day. These AIs are lower than the U.S RDAs. In both systems, teenagers need more than adults.[101] The European Food Safety Authority reviewed the same safety question and decided that there was not sufficient information to set a UL.[102]
For U.S. food and dietary supplement labeling purposes the amount in a serving is expressed as a percent of Daily Value (%DV). For phosphorus labeling purposes 100% of the Daily Value was 1000 mg, but as of May 27, 2016 it was revised to 1250 mg to bring it into agreement with the RDA.[103][104] A table of the old and new adult daily values is provided at Reference Daily Intake.
Food sources[edit]
The main food sources for phosphorus are the same as those containing protein, although proteins do not contain phosphorus. For example, milk, meat, and soya typically also have phosphorus. As a rule, if a diet has sufficient protein and calcium, the amount of phosphorus is probably sufficient.[105]
Precautions[edit]
Organic compounds of phosphorus form a wide class of materials; many are required for life, but some are extremely toxic. Fluorophosphate esters are among the most potent neurotoxins known. A wide range of organophosphorus compounds are used for their toxicity as pesticides (herbicides, insecticides, fungicides, etc.) and weaponised as nerve agents against enemy humans. Most inorganic phosphates are relatively nontoxic and essential nutrients.[15]
The white phosphorus allotrope presents a significant hazard because it ignites in air and produces phosphoric acid residue. Chronic white phosphorus poisoning leads to necrosis of the jaw called «phossy jaw». White phosphorus is toxic, causing severe liver damage on ingestion and may cause a condition known as «Smoking Stool Syndrome».[106]
In the past, external exposure to elemental phosphorus was treated by washing the affected area with 2% copper sulfate solution to form harmless compounds that are then washed away. According to the recent US Navy’s Treatment of Chemical Agent Casualties and Conventional Military Chemical Injuries: FM8-285: Part 2 Conventional Military Chemical Injuries, «Cupric (copper(II)) sulfate has been used by U.S. personnel in the past and is still being used by some nations. However, copper sulfate is toxic and its use will be discontinued. Copper sulfate may produce kidney and cerebral toxicity as well as intravascular hemolysis.»[107]
The manual suggests instead «a bicarbonate solution to neutralise phosphoric acid, which will then allow removal of visible white phosphorus. Particles often can be located by their emission of smoke when air strikes them, or by their phosphorescence in the dark. In dark surroundings, fragments are seen as luminescent spots. Promptly debride the burn if the patient’s condition will permit removal of bits of WP (white phosphorus) that might be absorbed later and possibly produce systemic poisoning. DO NOT apply oily-based ointments until it is certain that all WP has been removed. Following complete removal of the particles, treat the lesions as thermal burns.»[note 1][citation needed] As white phosphorus readily mixes with oils, any oily substances or ointments are not recommended until the area is thoroughly cleaned and all white phosphorus removed.
People can be exposed to phosphorus in the workplace by inhalation, ingestion, skin contact, and eye contact. The Occupational Safety and Health Administration (OSHA) has set the phosphorus exposure limit (Permissible exposure limit) in the workplace at 0.1 mg/m3 over an 8-hour workday. The National Institute for Occupational Safety and Health (NIOSH) has set a Recommended exposure limit (REL) of 0.1 mg/m3 over an 8-hour workday. At levels of 5 mg/m3, phosphorus is immediately dangerous to life and health.[108]
US DEA List I status[edit]
Phosphorus can reduce elemental iodine to hydroiodic acid, which is a reagent effective for reducing ephedrine or pseudoephedrine to methamphetamine.[109] For this reason, red and white phosphorus were designated by the United States Drug Enforcement Administration as List I precursor chemicals under 21 CFR 1310.02 effective on November 17, 2001.[110] In the United States, handlers of red or white phosphorus are subject to stringent regulatory controls.[110][111][112]
See also[edit]
- Phosphorus cycle
Notes[edit]
- ^ WP, (white phosphorus), exhibits chemoluminescence upon exposure to air and if there is any WP in the wound, covered by tissue or fluids such as blood serum, it will not glow until it is exposed to air, which requires a very dark room and dark-adapted eyes to see clearly
References[edit]
- ^ «Standard Atomic Weights: Phosphorus». CIAAW. 2013.
- ^ «Phosphorus: Chemical Element». Encyclopædia Britannica.
- ^ Wang, Yuzhong; Xie, Yaoming; Wei, Pingrong; King, R. Bruce; Schaefer, Iii; Schleyer, Paul v. R.; Robinson, Gregory H. (2008). «Carbene-Stabilized Diphosphorus». Journal of the American Chemical Society. 130 (45): 14970–1. doi:10.1021/ja807828t. PMID 18937460.
- ^ Ellis, Bobby D.; MacDonald, Charles L. B. (2006). «Phosphorus(I) Iodide: A Versatile Metathesis Reagent for the Synthesis of Low Oxidation State Phosphorus Compounds». Inorganic Chemistry. 45 (17): 6864–74. doi:10.1021/ic060186o. PMID 16903744.
- ^ Lide, D. R., ed. (2005). «Magnetic susceptibility of the elements and inorganic compounds». CRC Handbook of Chemistry and Physics (PDF) (86th ed.). Boca Raton (FL): CRC Press. ISBN 0-8493-0486-5.
- ^ Weast, Robert (1984). CRC, Handbook of Chemistry and Physics. Boca Raton, Florida: Chemical Rubber Company Publishing. pp. E110. ISBN 0-8493-0464-4.
- ^ cf. «Memoir on Combustion in General» Mémoires de l’Académie Royale des Sciences 1777, 592–600. from Henry Marshall Leicester and Herbert S. Klickstein, A Source Book in Chemistry 1400–1900 (New York: McGraw Hill, 1952)
- ^ a b A. Holleman; N. Wiberg (1985). «XV 2.1.3». Lehrbuch der Anorganischen Chemie (33rd ed.). de Gruyter. ISBN 3-11-012641-9.
- ^ a b Abundance. ptable.com
- ^ Simon, Arndt; Borrmann, Horst; Horakh, Jörg (1997). «On the Polymorphism of White Phosphorus». Chemische Berichte. 130 (9): 1235–1240. doi:10.1002/cber.19971300911.
- ^ a b Cossairt, Brandi M.; Cummins, Christopher C.; Head, Ashley R.; Lichtenberger, Dennis L.; Berger, Raphael J. F.; Hayes, Stuart A.; Mitzel, Norbert W.; Wu, Gang (2010-06-01). «On the Molecular and Electronic Structures of AsP3 and P4». Journal of the American Chemical Society. 132 (24): 8459–8465. doi:10.1021/ja102580d. ISSN 0002-7863. PMID 20515032.
- ^ Welford C. Roberts; William R. Hartley (1992-06-16). Drinking Water Health Advisory: Munitions (illustrated ed.). CRC Press, 1992. p. 399. ISBN 0873717546.
- ^ Marie-Thérèse Averbuch-Pouchot; A. Durif (1996). Topics in Phosphate Chemistry. World Scientific, 1996. p. 3. ISBN 9810226349.
- ^ Simon, Arndt; Borrmann, Horst; Horakh, Jörg (September 1997). «On the Polymorphism of White Phosphorus». Chemische Berichte. 130 (9): 1235–1240. doi:10.1002/cber.19971300911. ISSN 0009-2940.
- ^ a b c d e f g h i j k l m n Greenwood, N. N.; & Earnshaw, A. (1997). Chemistry of the Elements (2nd Edn.), Oxford:Butterworth-Heinemann. ISBN 0-7506-3365-4.
- ^ Piro, N. A.; Figueroa, J. S.; McKellar, J. T.; Cummins, C. C. (2006). «Triple-Bond Reactivity of Diphosphorus Molecules». Science. 313 (5791): 1276–9. Bibcode:2006Sci…313.1276P. doi:10.1126/science.1129630. PMID 16946068. S2CID 27740669.
- ^ a b c Berger, L. I. (1996). Semiconductor materials. CRC Press. p. 84. ISBN 0-8493-8912-7.
- ^ Shen, Z; Yu, JC (2016). «Nanostructured elemental photocatalysts: Development and challenges». In Yamashita, H; Li, H (eds.). Nanostructured Photocatalysts: Advanced Functional Materials. Switzerland: Springer. pp. 295–312 (301). ISBN 978-3-319-26077-8.
- ^ a b c d e Parkes & Mellor 1939, p. 717
- ^ a b Egon Wiberg; Nils Wiberg; Arnold Frederick Holleman (2001). Inorganic chemistry. Academic Press. pp. 683–684, 689. ISBN 978-0-12-352651-9. Retrieved 2011-11-19.
- ^ Parkes & Mellor 1939, pp. 721–722
- ^ a b c d Hammond, C. R. (2000). The Elements, in Handbook of Chemistry and Physics (81st ed.). CRC press. ISBN 0-8493-0481-4.
- ^ A. Brown; S. Runquist (1965). «Refinement of the crystal structure of black phosphorus». Acta Crystallogr. 19 (4): 684–685. doi:10.1107/S0365110X65004140.
- ^ Cartz, L.; Srinivasa, S.R.; Riedner, R.J.; Jorgensen, J.D.; Worlton, T.G. (1979). «Effect of pressure on bonding in black phosphorus». Journal of Chemical Physics. 71 (4): 1718–1721. Bibcode:1979JChPh..71.1718C. doi:10.1063/1.438523.
- ^ Lange, Stefan; Schmidt, Peer & Nilges, Tom (2007). «Au3SnP7@Black Phosphorus: An Easy Access to Black Phosphorus». Inorg. Chem. 46 (10): 4028–35. doi:10.1021/ic062192q. PMID 17439206.
- ^ Robert Engel (2003-12-18). Synthesis of Carbon-Phosphorus Bonds (2 ed.). CRC Press, 2003. p. 11. ISBN 0203998243.
- ^ «Nobel Prize in Chemistry 1956 – Presentation Speech by Professor A. Ölander (committee member)». Retrieved 2009-05-05.
- ^ «Phosphorus Topics page, at Lateral Science». Archived from the original on 2009-02-21. Retrieved 2009-05-05.
- ^ a b c Emsley, John (2000). The Shocking History of Phosphorus. London: Macmillan. ISBN 0-330-39005-8.
- ^ Vanzee, Richard J.; Khan, Ahsan U. (1976). «The phosphorescence of phosphorus». The Journal of Physical Chemistry. 80 (20): 2240–2242. doi:10.1021/j100561a021.
- ^ a b Michael A. Sommers (2007-08-15). Phosphorus. The Rosen Publishing Group, 2007. p. 25. ISBN 978-1404219601.
- ^ Audi, G.; Kondev, F. G.; Wang, M.; Huang, W. J.; Naimi, S. (2017). «The NUBASE2016 evaluation of nuclear properties» (PDF). Chinese Physics C. 41 (3): 030001. Bibcode:2017ChPhC..41c0001A. doi:10.1088/1674-1137/41/3/030001.
- ^ Neufcourt, L.; Cao, Y.; Nazarewicz, W.; Olsen, E.; Viens, F. (2019). «Neutron drip line in the Ca region from Bayesian model averaging». Physical Review Letters. 122 (6): 062502–1–062502–6. arXiv:1901.07632. Bibcode:2019PhRvL.122f2502N. doi:10.1103/PhysRevLett.122.062502. PMID 30822058. S2CID 73508148.
- ^ «Phosphorus-32» (PDF). University of Michigan Department of Occupational Safety & Environmental Health. Archived from the original (PDF) on 2016-05-28. Retrieved 2010-11-18.
- ^ Koo, B.-C.; Lee, Y.-H.; Moon, D.-S.; Yoon, S.-C.; Raymond, J. C. (2013). «Phosphorus in the Young Supernova Remnant Cassiopeia A». Science. 342 (6164): 1346–8. arXiv:1312.3807. Bibcode:2013Sci…342.1346K. doi:10.1126/science.1243823. PMID 24337291. S2CID 35593706.
- ^ Rivilla, V. M.; Drozdovskaya, M. N.; Altwegg, K.; Caselli, P.; Beltrán, M. T.; Fontani, F.; van der Tak, F. F. S.; Cesaroni, R.; Vasyunin, A.; Rubin, M.; Lique, F.; Marinakis, S.; Testi, L. (2019). «ALMA and ROSINA detections of phosphorus-bearing molecules: the interstellar thread between star-forming regions and comets». Monthly Notices of the Royal Astronomical Society. 492: 1180–1198. arXiv:1911.11647. doi:10.1093/mnras/stz3336. S2CID 208290964.
- ^ ESO (15 January 2020). «Astronomers reveal interstellar thread of one of life’s building blocks». Phys.org. Retrieved 16 January 2020.
- ^ «Phosphate Rock: Statistics and Information». USGS. Retrieved 2009-06-06.
- ^ a b c d e Philpott, Tom (March–April 2013). «You Need Phosphorus to Live—and We’re Running Out». Mother Jones.
- ^ Klein, Cornelis and Cornelius S. Hurlbut, Jr., Manual of Mineralogy, Wiley, 1985, 20th ed., p. 360, ISBN 0-471-80580-7
- ^ Threlfall 1951, p. 51
- ^ Arthur D. F. Toy (2013-10-22). The Chemistry of Phosphorus. Elsevier, 2013. p. 389. ISBN 978-1483147413.
- ^ D. E. C. Corbridge «Phosphorus: An Outline of its Chemistry, Biochemistry, and Technology» 5th Edition Elsevier: Amsterdam 1995. ISBN 0-444-89307-5.
- ^ Kutzelnigg, W. (1984). «Chemical Bonding in Higher Main Group Elements» (PDF). Angew. Chem. Int. Ed. Engl. 23 (4): 272–295. doi:10.1002/anie.198402721. Archived from the original (PDF) on 2020-04-16. Retrieved 2009-05-24.
- ^ Mark, J. E.; Allcock, H. R.; West, R. «Inorganic Polymers» Prentice Hall, Englewood, NJ: 1992. ISBN 0-13-465881-7.
- ^ Heal, H. G. «The Inorganic Heterocyclic Chemistry of Sulfur, Nitrogen, and Phosphorus» Academic Press: London; 1980. ISBN 0-12-335680-6.
- ^ Weeks, Mary Elvira (1932). «The discovery of the elements. II. Elements known to the alchemists». Journal of Chemical Education. 9 (1): 11. Bibcode:1932JChEd…9…11W. doi:10.1021/ed009p11.
- ^ Beatty, Richard (2000). Phosphorus. Marshall Cavendish. p. 7. ISBN 0-7614-0946-7.
- ^ a b Schmundt, Hilmar (21 April 2010), «Experts Warn of Impending Phosphorus Crisis», Der Spiegel.
- ^ Stillman, J. M. (1960). The Story of Alchemy and Early Chemistry. New York: Dover. pp. 418–419. ISBN 0-7661-3230-7.
- ^ Baccini, Peter; Paul H. Brunner (2012-02-10). Metabolism of the Anthroposphere. MIT Press, 2012. p. 288. ISBN 978-0262300544.
- ^ Emsley, John (7 January 2002). The 13th Element: The Sordid Tale of Murder, Fire, and Phosphorus. John Wiley & Sons. ISBN 978-0-471-44149-6. Retrieved 3 February 2012.
- ^ cf. «Memoir on Combustion in General» Mémoires de l’Académie Royale des Sciences 1777, 592–600. from Henry Marshall Leicester and Herbert S. Klickstein, A Source Book in Chemistry 1400–1900 (New York: McGraw Hill, 1952)
- ^ Thomson, Robert Dundas (1870). Dictionary of chemistry with its applications to mineralogy, physiology and the arts. Rich. Griffin and Company. p. 416.
- ^ Threlfall 1951, pp. 49–66
- ^ Robert B. Heimann; Hans D. Lehmann (2015-03-10). Bioceramic Coatings for Medical Implants. John Wiley & Sons, 2015. p. 4. ISBN 978-3527684007.
- ^ The Chemistry of Phosphorus, by Arthur Toy
- ^ US patent 417943
- ^ Threlfall 1951, pp. 81–101
- ^ Parkes & Mellor 1939, p. 718–720.
- ^ a b c Threlfall 1951, pp. 167–185
- ^ Lewis R. Goldfrank; Neal Flomenbaum; Mary Ann Howland; Robert S. Hoffman; Neal A. Lewin; Lewis S. Nelson (2006). Goldfrank’s toxicologic emergencies. McGraw-Hill Professional. pp. 1486–1489. ISBN 0-07-143763-0.
- ^ The White Phosphorus Matches Prohibition Act, 1908.
- ^ «Phosphate Rock» (PDF). USGS. Retrieved 2017-03-20.
- ^ Lewis, Leo (2008-06-23). «Scientists warn of lack of vital phosphorus as biofuels raise demand». The Times.
- ^ Grantham, Jeremy (Nov 12, 2012). «Be persuasive. Be brave. Be arrested (if necessary)». Nature. 491 (7424): 303. Bibcode:2012Natur.491..303G. doi:10.1038/491303a. PMID 23151541.
- ^ a b Geeson, Michael B.; Cummins, Christopher C. (2020). «Let’s Make White Phosphorus Obsolete». ACS Central Science. 6 (6): 848–860. doi:10.1021/acscentsci.0c00332. PMC 7318074. PMID 32607432.
- ^ Tayibi, Hanan; Choura, Mohamed; López, Félix A.; Alguacil, Francisco J.; López-Delgado, Aurora (2009). «Environmental Impact and Management of Phosphogypsum». Journal of Environmental Management. 90 (8): 2377–2386. doi:10.1016/j.jenvman.2009.03.007. hdl:10261/45241. PMID 19406560.
- ^ Shriver, Atkins. Inorganic Chemistry, Fifth Edition. W. H. Freeman and Company, New York; 2010; p. 379.
- ^ «ERCO and Long Harbour». Memorial University of Newfoundland and the C.R.B. Foundation. Retrieved 2009-06-06.
- ^ Von Wagner, Rudolf (1897). Manual of chemical technology. New York: D. Appleton & Co. p. 411.
- ^ «Current EU approved additives and their E Numbers». Foods Standards Agency. 14 March 2012. Archived from the original on 21 August 2013. Retrieved 22 July 2012.
- ^ «Why is phosphoric acid used in some Coca‑Cola drinks?| Frequently Asked Questions | Coca-Cola GB». www.coca-cola.co.uk. Archived from the original on 2 August 2021. Retrieved 2021-08-31.
- ^ Moynihan, P. J. (23 November 2002). «Dietary advice in dental practice». British Dental Journal. 193 (10): 563–568. doi:10.1038/sj.bdj.4801628. PMID 12481178.
- ^ Qaseem, A; Dallas, P; Forciea, MA; Starkey, M; et al. (4 November 2014). «Dietary and pharmacologic management to prevent recurrent nephrolithiasis in adults: A clinical practice guideline from the American College of Physicians». Annals of Internal Medicine. 161 (9): 659–67. doi:10.7326/M13-2908. PMID 25364887.
- ^ a b Etesami, H. (2019). Nutrient Dynamics for Sustainable Crop Production. Springer. p. 217. ISBN 9789811386602.
- ^ «Soil Phosphorous» (PDF). United States Department of Agriculture.
- ^ «Managing Phosphorus for Crop Production». Penn State Extension. Archived from the original on 2020-10-20. Retrieved 2020-08-17.
- ^ Jessica Elzea Kogel, ed. (2006). Industrial Minerals & Rocks: Commodities, Markets, and Uses. SME, 2006. p. 964. ISBN 0873352335.
- ^ a b c d e Threlfall, R.E. (1951). 100 years of Phosphorus Making: 1851–1951. Oldbury: Albright and Wilson Ltd.
- ^ Diskowski, Herbert and Hofmann, Thomas (2005) «Phosphorus» in Ullmann’s Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim. doi:10.1002/14356007.a19_505
- ^ Roland W. Scholz; Amit H. Roy; Fridolin S. Brand; Deborah Hellums; Andrea E. Ulrich, eds. (2014-03-12). Sustainable Phosphorus Management: A Global Transdisciplinary Roadmap. Springer Science & Business Media. p. 175. ISBN 978-9400772502.
- ^ Mel Schwartz (2016-07-06). Encyclopedia and Handbook of Materials, Parts and Finishes. CRC Press. ISBN 978-1138032064.
- ^ Joseph R. Davisz, ed. (January 2001). Copper and Copper Alloys. ASM International. p. 181. ISBN 0871707268.
- ^ Hughes, J. P. W; Baron, R.; Buckland, D. H.; et al. (1962). «Phosphorus Necrosis of the Jaw: A Present-day Study: With Clinical and Biochemical Studies». Br. J. Ind. Med. 19 (2): 83–99. doi:10.1136/oem.19.2.83. PMC 1038164. PMID 14449812.
- ^ Crass, M. F. Jr. (1941). «A history of the match industry. Part 9» (PDF). Journal of Chemical Education. 18 (9): 428–431. Bibcode:1941JChEd..18..428C. doi:10.1021/ed018p428.[permanent dead link]
- ^ Oliver, Thomas (1906). «Industrial disease due to certain poisonous fumes or gases». Archives of the Public Health Laboratory. Manchester University Press. 1: 1–21.
- ^ Charnovitz, Steve (1987). «The Influence of International Labour Standards on the World Trading Regime. A Historical Overview». International Labour Review. 126 (5): 565, 571.
- ^ Alexander P. Hardt (2001). «Matches». Pyrotechnics. Post Falls Idaho USA: Pyrotechnica Publications. pp. 74–84. ISBN 0-929388-06-2.
- ^ Klaus Schrödter, Gerhard Bettermann, Thomas Staffel, Friedrich Wahl, Thomas Klein, Thomas Hofmann «Phosphoric Acid and Phosphates» in Ullmann’s Encyclopedia of Industrial Chemistry 2008, Wiley-VCH, Weinheim. doi:10.1002/14356007.a19_465.pub3
- ^ «Obsolete hand grenades». GlobalSecurity.Org. Retrieved 2009-08-03.
- ^ Dockery, Kevin (1997). Special Warfare Special Weapons. Chicago: Emperor’s Press. ISBN 1-883476-00-3.
- ^ David A. Atwood, ed. (2013-02-19). Radionuclides in the Environment. John Wiley & Sons, 2013. ISBN 978-1118632697.
- ^ Ruttenberg, K. C. Phosphorus Cycle – Terrestrial Phosphorus Cycle, Transport of Phosphorus, from Continents to the Ocean, The Marine Phosphorus Cycle. (archived link).
- ^ Lipmann, D. (1944). «Enzymatic Synthesis of Acetyl Phosphate». J Biol Chem. 155: 55–70. doi:10.1016/S0021-9258(18)43172-9.
- ^ Nelson, D. L.; Cox, M. M. «Lehninger, Principles of Biochemistry» 3rd Ed. Worth Publishing: New York, 2000. ISBN 1-57259-153-6.
- ^ Bernhardt, Nancy E.; Kasko, Artur M. (2008). Nutrition for the Middle Aged and Elderly. Nova Publishers. p. 171. ISBN 978-1-60456-146-3.
- ^ Mehanna H. M., Moledina J., Travis J. (June 2008). «Refeeding syndrome: what it is, and how to prevent and treat it». BMJ. 336 (7659): 1495–8. doi:10.1136/bmj.a301. PMC 2440847. PMID 18583681.
{{cite journal}}: CS1 maint: uses authors parameter (link) - ^ Anderson, John J. B. (1996). «Calcium, Phosphorus and Human Bone Development». Journal of Nutrition. 126 (4 Suppl): 1153S–1158S. doi:10.1093/jn/126.suppl_4.1153S. PMID 8642449.
- ^ Institute of Medicine (1997). «Phosphorus». Dietary Reference Intakes for Calcium, Phosphorus, Magnesium, Vitamin D, and Fluoride. Washington, DC: The National Academies Press. pp. 146–189. doi:10.17226/5776. ISBN 978-0-309-06403-3. PMID 23115811. S2CID 8768378.
- ^ «Overview on Dietary Reference Values for the EU population as derived by the EFSA Panel on Dietetic Products, Nutrition and Allergies» (PDF). 2017.
- ^ Tolerable Upper Intake Levels For Vitamins And Minerals (PDF), European Food Safety Authority, 2006
- ^ «Federal Register May 27, 2016 Food Labeling: Revision of the Nutrition and Supplement Facts Labels. FR page 33982» (PDF).
- ^ «Daily Value Reference of the Dietary Supplement Label Database (DSLD)». Dietary Supplement Label Database (DSLD). Archived from the original on 7 April 2020. Retrieved 16 May 2020.
- ^ Phosphorus in diet: MedlinePlus Medical Encyclopedia. Nlm.nih.gov (2011-11-07). Retrieved on 2011-11-19.
- ^ «CBRNE – Incendiary Agents, White Phosphorus (Smoking Stool Syndrome)». Retrieved 2009-05-05.
- ^ «US Navy’s Treatment of Chemical Agent Casualties and Conventional Military Chemical Injuries: FM8-285: Part 2 Conventional Military Chemical Injuries». Archived from the original on November 22, 2005. Retrieved 2009-05-05.
- ^ «CDC — NIOSH Pocket Guide to Chemical Hazards — Phosphorus (yellow)». www.cdc.gov. Retrieved 2015-11-21.
- ^ Skinner, H.F. (1990). «Methamphetamine synthesis via hydriodic acid/red phosphorus reduction of ephedrine». Forensic Science International. 48 (2): 123–134. doi:10.1016/0379-0738(90)90104-7.
- ^ a b «66 FR 52670—52675». 17 October 2001. Retrieved 2009-05-05.
- ^ «21 cfr 1309». Archived from the original on 2009-05-03. Retrieved 2009-05-05.
- ^ «21 USC, Chapter 13 (Controlled Substances Act)». Retrieved 2009-05-05.
Bibliography[edit]
- Emsley, John (2000). The Shocking history of Phosphorus. A biography of the Devil’s Element. London: MacMillan. ISBN 0-333-76638-5.
- Parkes, G. D.; Mellor, J. W. (1939). Mellor’s Modern Inorganic Chemistry. Longman’s Green and Co.
- Podger, Hugh (2002). Albright & Wilson. The Last 50 years. Studley: Brewin Books. ISBN 1-85858-223-7.
- Threlfall, Richard E. (1951). The Story of 100 years of Phosphorus Making: 1851–1951. Oldbury: Albright & Wilson Ltd.
| Фосфор | |
|---|---|

Белый, красный и черный фосфор |
|
| Название, символ, номер | Фосфор/ Phosphorus (P), 15 |
| Атомная масса (молярная масса) |
30,973762(2) а. е. м. (г/моль) |
| Электронная конфигурация | [Ne] 3s2 3p3 |
| Радиус атома | 128 пм |
| Ковалентный радиус | 106 пм |
| Радиус иона | 35 (+5e) 212 (-3e) пм |
| Электроотрицательность | 2,19 (шкала Полинга) |
| Электродный потенциал | 0 |
| Степени окисления | 5, 3, 1, 0, −1, −3 |
| Энергия ионизации (первый электрон) |
1011,2(10,48) кДж/моль (эВ) |
| Плотность (при н. у.) | (белый фосфор)1,82 г/см³ |
| Температура плавления | 44,15 °C (317,3 K) |
| Температура кипения | 279,85 °C (553 K) |
| Уд. теплота плавления | 2,51 кДж/моль |
| Уд. теплота испарения | 49,8 кДж/моль |
| Молярная теплоёмкость | 21,6 (ромбич.) Дж/(K·моль) |
| Молярный объём | 17,0 см³/моль |
| Структура решётки | кубическая, объёмноцентрированная |
| Параметры решётки | 18,800 Å |
| Теплопроводность | (300 K) (0,236) Вт/(м·К) |
| Номер CAS | 7723-14-0 |
Фосфор (от др.-греч. φῶς — свет и φέρω — несу; φωσφόρος — светоносный; лат. Phosphorus) — химический элемент 15-й группы (по устаревшей классификации — главной подгруппы пятой группы) третьего периода периодической системы Д. И. Менделеева; имеет атомный номер 15. Элемент входит в группу пниктогенов. Фосфор — один из распространённых элементов земной коры: его содержание составляет 0,08—0,09 % её массы. Концентрация в морской воде 0,07 мг/л. В свободном состоянии не встречается из-за высокой химической активности. Образует около 190 минералов, важнейшими из которых являются апатит Ca5(PO4)3 (F,Cl,OH), фосфорит (Сa3(PO4)2) и другие. Фосфор входит в состав важнейших биологических соединений — фосфолипидов. Содержится в животных тканях, входит в состав белков и других важнейших органических соединений (АТФ, ДНК), является элементом жизни.
Содержание
- 1 История
- 2 Происхождение названия
- 3 Получение
- 4 Физические свойства
- 4.1 Белый фосфор
- 4.2 Жёлтый фосфор
- 4.3 Красный фосфор
- 4.4 Чёрный фосфор
- 4.5 Металлический фосфор
- 5 Химические свойства
- 5.1 Взаимодействие с простыми веществами
- 5.2 Взаимодействие с водой
- 5.3 Взаимодействие со щелочами
- 5.4 Восстановительные свойства
- 6 Изотопы
- 7 Применение
- 7.1 Элементарный фосфор
- 7.2 Соединения фосфора в сельском хозяйстве
- 7.3 Соединения фосфора в промышленности
- 7.4 Фосфатные связующие
- 8 Биологическая роль соединений фосфора
- 8.1 Токсикология элементарного фосфора
- 8.2 Токсикология соединений фосфора
- 9 Опасность для здоровья

История
Фосфор открыт гамбургским алхимиком Хеннигом Брандом в 1669 году. Подобно другим алхимикам, Бранд пытался отыскать философский камень, а получил светящееся вещество. Бранд сфокусировался на опытах с человеческой мочой, так как полагал, что она, обладая золотистым цветом, может содержать золото или нечто нужное для его добычи. Первоначально его способ заключался в том, что сначала моча отстаивалась в течение нескольких дней, пока не исчезнет неприятный запах, а затем кипятилась до клейкого состояния. Нагревая эту пасту до высоких температур и доводя до появления пузырьков, он надеялся, что, сконденсировавшись, они будут содержать золото. После нескольких часов интенсивных кипячений получались крупицы белого воскоподобного вещества, которое очень ярко горело и к тому же мерцало в темноте. Бранд назвал это вещество phosphorus mirabilis (лат. «чудотворный носитель света»). Открытие фосфора Брандом стало первым открытием нового элемента со времён античности.

Картина Джозефа Райта «Алхимик, открывающий фосфор» (1771 год), предположительно описывающая открытие фосфора Хеннигом Брандом.
Несколько позже фосфор был получен другим немецким химиком — Иоганном Кункелем.
Независимо от Бранда и Кункеля фосфор был получен Р. Бойлем, описавшим его в статье «Способ приготовления фосфора из человеческой мочи», датированной 14 октября 1680 года и опубликованной в 1693 году.
Более усовершенствованный способ получения фосфора был опубликован в 1743 году Андреасом Маргграфом.
Существуют данные, что фосфор умели получать ещё арабские алхимики в XII в.
То, что фосфор — простое вещество, доказал Лавуазье.
Аморфную аллотропную модификацию фосфора — красный фосфор Pn — выделил, нагревая белый фосфор без доступа воздуха, А. Шрёттер в середине XIX в.
Происхождение названия
В 1669 году Хеннинг Бранд при нагревании смеси белого песка и выпаренной мочи получил светящееся в темноте вещество, названное сначала «холодным огнём». Вторичное название «фосфор» происходит от греческих слов «φώς» — свет и «φέρω» — несу. В древнегреческой мифологии имя Фосфор (или Эосфор, др.-греч. Φωσφόρος) носил страж Утренней звезды.
Получение
Фосфор получают из апатитов или фосфоритов в результате взаимодействия с коксом и кремнезёмом при температуре около 1600 °С:
-
- 2Ca3(PO4)2 + 10C + 6SiO2 → P4 + 10CO + 6CaSiO3 или Ca3(PO4)2 + 3SiO2+5C = 3CaSiO3+5CO+2P.
Образующиеся пары фосфора конденсируются в приёмнике под слоем воды в аллотропическую модификацию в виде белого фосфора. Вместо фосфоритов для получения элементарного фосфора можно восстанавливать углём и другие неорганические соединения фосфора, например, в том числе, метафосфорную кислоту:
-
- 4HPO3 + 10C → P4 + 2H2O + 10CO
Физические свойства
Элементарный фосфор при нормальных условиях существует в виде нескольких устойчивых аллотропических модификаций. Все существующие аллотропные модификации фосфора пока (2016 г.) до конца не изучены. Традиционно различают четыре его модификации: зеленовато-белый, красный, чёрный и металлический фосфор. Иногда их ещё называют главными аллотропными модификациями, подразумевая при этом, что все остальные описываемые модификации являются смесью этих четырёх. При стандартных условиях устойчивы только три аллотропических модификации фосфора (например, белый фосфор термодинамически неустойчив (квазистационарное состояние) и переходит со временем при нормальных условиях в красный фосфор). В условиях сверхвысоких давлений термодинамически устойчива металлическая форма элемента. Все модификации различаются по цвету, плотности и другим физическим и химическим характеристикам, особенно по химической активности. При переходе состояния вещества в более термодинамически устойчивую модификацию снижается химическая активность, например, при последовательном превращении белого фосфора в красный, потом красного в чёрный (металлический).

Аллотропные модификации фосфора (белый, красный, чёрный)
Белый фосфор
Белый фосфор представляет собой белое вещество (из-за примесей может иметь желтоватый оттенок). По внешнему виду он очень похож на очищенный воск или парафин, легко режется ножом и деформируется от небольших усилий.
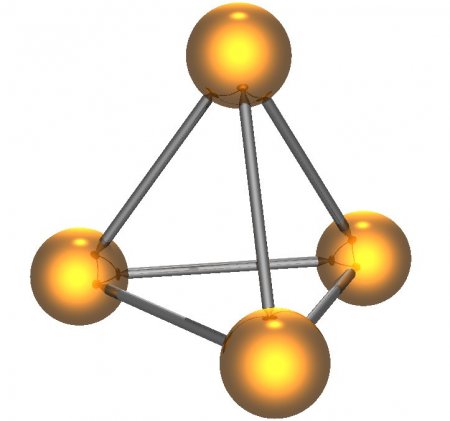
Белый фосфор имеет молекулярную кристаллическую решётку, формула молекулы белого фосфора — P4, причём атомы расположены в вершинах тетраэдра. Отливаемый в инертной атмосфере в виде палочек (слитков), он сохраняется в отсутствии воздуха под слоем очищенной воды или в специальных инертных средах.
Плохо растворяется в воде, но легкорастворим в органических растворителях. Растворимостью белого фосфора в сероуглероде пользуются для промышленной очистки его от примесей. Плотность белого фосфора из всех его модификаций наименьшая и составляет около 1823 кг/м³. Плавится белый фосфор при 44,1 °C. В парообразном состоянии происходит диссоциация молекул фосфора.
Химически белый фосфор чрезвычайно активен. Например, он медленно окисляется кислородом воздуха уже при комнатной температуре и светится (бледно-зелёное свечение). Явление такого рода свечения вследствие химических реакций окисления называется хемилюминесценцией (иногда ошибочно фосфоресценцией). При взаимодействии с кислородом белый фосфор горит даже под водой.
Белый фосфор не только активен химически, но и весьма ядовит: летальная доза белого фосфора для взрослого человека составляет 0,05—0,15 г, а при хроническом отравлении поражает кости, например, вызывает омертвение челюстей. При контакте с кожей легко самовоспламеняется, вызывая серьёзные ожоги.
Под действием света, при нагревании до не очень высоких температур в безвоздушной среде, а также под действием ионизирующего излучения белый фосфор превращается в красный фосфор.
Жёлтый фосфор
Неочищенный белый фосфор обычно называют «жёлтый фосфор». Сильно ядовитое (ПДК в атмосферном воздухе 0,0005 мг/м³), огнеопасное кристаллическое вещество от светло-жёлтого до тёмно-бурого цвета. Плотность 1,83 г/см³, плавится при +43,1 °C, кипит при +280 °C. В воде не растворяется, на воздухе легко окисляется и самовоспламеняется. Горит ослепительным ярко-зелёным пламенем с выделением густого белого дыма — мелких частичек декаоксида тетрафосфора P4O10.
Так как фосфор реагирует с водой лишь при температуре свыше 500 °C, то для тушения фосфора используют воду в больших количествах (для снижения температуры очага возгорания и перевода фосфора в твёрдое состояние) или раствор сульфата меди (медного купороса), после гашения фосфор засыпают влажным песком. Для предохранения от самовозгорания жёлтый фосфор хранится и перевозится под слоем воды (раствора хлорида кальция).
Красный фосфор

Красный фосфор — это более термодинамически стабильная модификация элементарного фосфора. Впервые он был получен в 1847 году в Швеции австрийским химиком А. Шрёттером при нагревании белого фосфора при 500 °С в атмосфере угарного газа (СО) в запаянной стеклянной ампуле.
Красный фосфор имеет формулу Рn и представляет собой полимер со сложной структурой. В зависимости от способа получения и степени дробления, красный фосфор имеет оттенки от пурпурно-красного до фиолетового, а в литом состоянии — тёмно-фиолетовый с медным оттенком, имеет металлический блеск. Химическая активность красного фосфора значительно ниже, чем у белого; ему присуща исключительно малая растворимость. Растворить красный фосфор возможно лишь в некоторых расплавленных металлах (свинец и висмут), чем иногда пользуются для получения крупных его кристаллов. Так, например, немецкий физико-химик И. В. Гитторф в 1865 году впервые получил прекрасно построенные, но небольшие по размеру кристаллы (фосфор Гитторфа). Красный фосфор на воздухе не самовоспламеняется, вплоть до температуры 240—250 °С (при переходе в белую форму во время возгонки), но самовоспламеняется при трении или ударе, у него полностью отсутствует явление хемилюминесценции. Нерастворим в воде, а также в бензоле, сероуглероде и других веществах, растворим в трибромиде фосфора. При температуре возгонки красный фосфор превращается в пар, при охлаждении которого образуется в основном белый фосфор.
Ядовитость его в тысячи раз меньше, чем у белого, поэтому он применяется гораздо шире, например, в производстве спичек (составом на основе красного фосфора покрыта тёрочная поверхность коробков). Плотность красного фосфора также выше, и достигает 2400 кг/м³ в литом виде. При хранении на воздухе красный фосфор в присутствии влаги постепенно окисляется, образуя гигроскопичный оксид, поглощает воду и отсыревает («отмокает»), образуя вязкую фосфорную кислоту; поэтому его хранят в герметичной таре. При «отмокании» — промывают водой от остатков фосфорных кислот, высушивают и используют по назначению.
Чёрный фосфор
Чёрный фосфор — это наиболее стабильная термодинамически и химически наименее активная форма элементарного фосфора. Впервые чёрный фосфор был получен в 1914 году американским физиком П. У. Бриджменом из белого фосфора в виде чёрных блестящих кристаллов, имеющих высокую (2690 кг/м³) плотность. Для проведения синтеза чёрного фосфора Бриджмен применил давление в 2⋅109 Па (20 тысяч атмосфер) и температуру около 200 °С. Начало быстрого перехода лежит в области 13 000 атмосфер и температуре около 230 °С.
Чёрный фосфор представляет собой чёрное вещество с металлическим блеском, жирное на ощупь и весьма похожее на графит, и с полностью отсутствующей растворимостью в воде или органических растворителях. Поджечь чёрный фосфор можно, только предварительно сильно раскалив в атмосфере чистого кислорода до 400 °С. Чёрный фосфор проводит электрический ток и имеет свойства полупроводника. Температура плавления чёрного фосфора 1000 °С под давлением 1,8⋅106 Па.
Металлический фосфор
При 8,3⋅1010 Па чёрный фосфор переходит в новую, ещё более плотную и инертную металлическую фазу с плотностью 3,56 г/см³, а при дальнейшем повышении давления до 1,25⋅1011 Па — ещё более уплотняется и приобретает кубическую кристаллическую решётку, при этом его плотность возрастает до 3,83 г/см³. Металлический фосфор очень хорошо проводит электрический ток.
Химические свойства
Химическая активность фосфора значительно выше, чем у азота. Химические свойства фосфора во многом определяются его аллотропной модификацией. Белый фосфор очень активен, в процессе перехода к красному и чёрному фосфору химическая активность снижается. Белый фосфор в воздухе при окислении кислородом воздуха при комнатной температуре излучает видимый свет, свечение обусловлено фотоэмиссионной реакцией окисления фосфора.
В жидком и растворенном состоянии, а также в парах до 800 °С фосфор состоит из молекул Р4. При нагревании выше 800 °С молекулы диссоциируют: Р4 = 2Р2. При температуре выше 2000 °С молекулы распадаются на атомы.
Взаимодействие с простыми веществами
Фосфор легко окисляется кислородом:
-
- 4P + 5O2 → 2P2O5 (с избытком кислорода)
- 4P + 3O2 → 2P2O3 (при медленном окислении или при недостатке кислорода)
Взаимодействует со многими простыми веществами — галогенами, серой, некоторыми металлами, проявляя окислительные и восстановительные свойства:
с металлами — окислитель, образует фосфиды:
-
- 2P + 3Ca → Ca3P2
фосфиды разлагаются водой и кислотами с образованием фосфина
с неметаллами — восстановитель:
-
- 2P + 3S → P2S3
- 2P + 5Cl2 → 2PCl5
С водородом фосфор практически не соединяется. Однако разложением некоторых фосфидов водой по реакции, например:
-
- Ca3P2 + 6H2O → 2PH3 + 3Ca(OH)2
может быть получен аналогичный аммиаку фосфористый водород (фосфин) — PH3
Взаимодействие с водой
Взаимодействует с водяным паром при температуре выше 500 °С, протекает реакция диспропорционирования с образованием фосфина и фосфорной кислоты:
-
- 8P + 12H2O →>500oC 5PH3 + 3H3PO4
Реакция взаимодействия красного фосфора и воды с образованием ортофосфорной кислоты и водорода. Реакция протекает при температуре 700—900 °C. Катализатором могут выступать: платина, медь, титан, цирконий.
-
- 2P + 8H2O →700−900oC,kat 2H3PO4 + 5H2
Взаимодействие со щелочами
В холодных концентрированных растворах щелочей также медленно протекает реакция диспропорционирования:
-
- 4P + 3KOH + 3H2O →τ PH3 + 3KH2PO2
Восстановительные свойства
Сильные окислители превращают фосфор в фосфорную кислоту:
-
- 3P + 5HNO3 + 2H2O → 3H3PO4 + 5NO
- 2P + 5H2SO4 → 2H3PO4 + 5SO2 + 2H2O
Реакция окисления фосфора происходит при поджигании спичек, в качестве окислителя выступает бертолетова соль:
-
- 6P + 5KClO3 → 5KCl + 3P2O5
Изотопы
Основная статья: Изотопы фосфора
Известно более 20 изотопов фосфора (с массовым числом от 24 до 47). Природный изотоп 31P стабилен. Из радиоактивных изотопов наиболее долгоживущие: 30P (T1/2 = 2,5 мин), 32P (T1/2 = 14,26 сут) и 33P (T1/2 = 25,34 сут)..
Применение
Фосфор является важнейшим биогенным элементом и в то же время находит очень широкое применение в промышленности. Красный фосфор применяют в производстве спичек. Его вместе с тонко измельчённым стеклом и клеем наносят на боковую поверхность коробки. При трении спичечной головки, в состав которой входят хлорат калия и сера, происходит воспламенение.
Элементарный фосфор
Пожалуй, первое свойство фосфора, которое человек поставил себе на службу, — это горючесть. Горючесть фосфора очень велика и зависит от аллотропической модификации.
Наиболее активен химически, токсичен и горюч белый («жёлтый») фосфор, потому он очень часто применяется (в зажигательных бомбах и пр.).
Красный фосфор — основная модификация, производимая и потребляемая промышленностью. Он применяется в производстве спичек, взрывчатых веществ, зажигательных составов, различных типов топлива, а также противозадирных смазочных материалов, в качестве газопоглотителя в производстве ламп накаливания.
Соединения фосфора в сельском хозяйстве
Фосфор (в виде фосфатов) — один из трёх важнейших биогенных элементов, участвует в синтезе АТФ. Большая часть производимой фосфорной кислоты идёт на получение фосфорных удобрений — суперфосфата, преципитата, аммофоски и др.
Соединения фосфора в промышленности
Фосфаты широко используются:
- в качестве комплексообразователей (средства для умягчения воды),
- в составе пассиваторов поверхности металлов (защита от коррозии, например, т. н. состав «мажеф»).
Фосфатные связующие
Способность фосфатов формировать прочную трёхмерную полимерную сетку используется для изготовления фосфатных и алюмофосфатных связок.
Черный (металлический) фосфор
Биологическая роль соединений фосфора
Фосфор присутствует в живых клетках в виде орто- и пирофосфорной кислот, входит в состав нуклеотидов, нуклеиновых кислот, фосфопротеидов, фосфолипидов, коферментов, ферментов. Кости человека состоят из гидроксилапатита 3Ca3(PO4)2·Ca(OH)2. В состав зубной эмали входит фторапатит. Основную роль в превращениях соединений фосфора в организме человека и животных играет печень. Обмен фосфорных соединений регулируется гормонами и витамином D. При недостатке фосфора в организме развиваются различные заболевания костей.
Суточная потребность в фосфоре составляет:
- для взрослых 1,0—2,0 г
- для беременных и кормящих женщин 3—3,8 г
- для детей и подростков 1,5—2,5 г
При больших физических нагрузках потребность в фосфоре возрастает в 1,5—2 раза.
Усвоение происходит эффективнее при приёме фосфора вместе с кальцием в соотношении 3:2 (P:Ca).
Некоторые источники:
| Продукт | Содержание, мг/100 г |
|---|---|
| Очищенное конопляное семя | 1650 |
| Семена тыквы (ядра) | 1233 |
| Семена подсолнечника (ядра) поджаренные | 1158 |
| Семена мака | 870 |
| Горчичный порошок | 828 |
| Кунжут (очищенный) | 774 |
| Семена дыни (ядра) | 755 |
| Какао-порошок | 734 |
| Твёрдый пармезан | 694 |
| Семена подсолнечника (ядра) сушёные | 660 |
| Сафлора семена (ядра) | 644 |
| Семена льна | 642 |
| Семена лотоса | 626 |
| Сыр швейцарский нежирный | 605 |
| Кешью сырые | 593 |
| Орехи пили | 575 |
| Амарантовая крупа | 557 |
| Сыр гауда | 546 |
| Овёс | 523 |
| Грецкий орех чёрный | 513 |
| Печень говяжья тушёная | 497 |
| Фисташки сырые | 490 |
| Миндаль | 481 |
| Киноа | 457 |
| Люпин, семена | 440 |
| Карп | 415 |
| Фасоль | 407 |
| Арахис | 397 |
| Сыр рокфор | 392 |
| Мука из цельного зерна | 357 |
| Печень куриная | 297 |
| Вырезка свиная | 286 |
| Желтоперый тунец | 278 |
| Сгущённое молоко | 253 |
| Яйцо | 198 |
| Говядина | 188 |
| Курица | 178 |
Токсикология элементарного фосфора
- Красный фосфор практически нетоксичен (токсичность ему придают примеси белого фосфора). Пыль красного фосфора, попадая в легкие, вызывает пневмонию при хроническом действии.
- Белый фосфор очень ядовит, растворим в липидах. Смертельная доза белого фосфора — 50—150 мг. Попадая на кожу, тлеющий белый фосфор даёт тяжелые ожоги.
Острые отравления фосфором проявляются жжением во рту и желудке, головной болью, слабостью, рвотой. Через 2—3 суток развивается желтуха. Для хронических форм характерны нарушение кальциевого обмена, поражение сердечно-сосудистой и нервной систем. Первая помощь при остром отравлении — промывание желудка, слабительное, очистительные клизмы, внутривенно растворы глюкозы. При ожогах кожи обработать поражённые участки растворами медного купороса или соды. ПДК паров фосфора в воздухе производственных помещений — 0,03 мг/м³, временно допустимая концентрация в атмосферном воздухе — 0,0005 мг/м³, ПДК в питьевой воде — 0,0001 мг/дм³.
Токсикология соединений фосфора
Некоторые соединения фосфора (фосфин) очень токсичны. Ввиду высокой (ЛД50 15-100 мг/кг) и чрезвычайно высокой (<15 мг/кг) токсичности большинство фосфорорганических соединений (ФОС) используются в качестве пестицидов (инсектициды, акарициды, зооциды и т. д.) или боевых отравляющих веществ. Примером боевых отравляющих веществ являются — зарин, зоман, табун, новичок, V-газы.
ФОС проявляют свойства веществ нервно-паралитического действия. Токсичность фосфорорганических соединений обусловлена ингибированием фермента ацетилхолинэстеразы, вследствие чего развивается головная боль, тошнота, головокружение, сужение зрачков (миоз), затруднение дыхания (отдышка), возникает слюнотечение, понижается артериальное давление, возникают конвульсии, проявляется паралитическое воздействие, кома, и как следствие может быстро возникнуть летальный исход. Эффективным антидотом при отравлении ФОС является атропин.
Опасность для здоровья
Рейтинг NFPA 704:
Фосфор относится к 1-му классу опасности.
Считается, что фосфор — это светящийся в темноте минерал, ядовитый и огнеопасный. Но это только часть правды об этом удивительном элементе. Фосфор бывает и иным, с прямо противоположными свойствами.
Что такое красный фосфор?
Фосфор может существовать в нескольких вариантах  (аллотропических формах), которые сильно отличаются своими физическими и химическими свойствами. Причиной этого являются различия в строении. Например, кристаллическая решетка белого фосфора молекулярная, а решетка красного фосфора — атомная. Благодаря ей он медленно реагирует с другими веществами, стабилен на воздухе в обычных условиях (белый фосфор на воздухе воспламеняется). Всего у фосфора найдено более двадцати модификаций, четыре из которых стабильны (белый, красный, черный и металлический фосфор), остальные — нестабильны.
(аллотропических формах), которые сильно отличаются своими физическими и химическими свойствами. Причиной этого являются различия в строении. Например, кристаллическая решетка белого фосфора молекулярная, а решетка красного фосфора — атомная. Благодаря ей он медленно реагирует с другими веществами, стабилен на воздухе в обычных условиях (белый фосфор на воздухе воспламеняется). Всего у фосфора найдено более двадцати модификаций, четыре из которых стабильны (белый, красный, черный и металлический фосфор), остальные — нестабильны.
Красный фосфор представляет собой очень интересное вещество, естественный неорганический полимер с формулой (Р4)n и весьма сложной структурой из пирамидально связанных атомов.
Свойства красного фосфора в некоторой степени зависят от условий его получения. Изменяя температуру, свет и катализаторы, можно создавать виды красного фосфора с прогнозируемыми свойствами.
Первооткрывателем красного фосфора является австриец А.Шрёттер, который получил его, нагревая запаянную ампулу с белым фосфором и угарным газом при температуре +500 °С.
Свойства красного фосфора
 Красный фосфор получают методом продолжительного нагревания белого фосфора при высоких температурах (250-300 °С) без доступа воздуха. Цвет вещества варьируется от пурпурно-красного до фиолетового.
Красный фосфор получают методом продолжительного нагревания белого фосфора при высоких температурах (250-300 °С) без доступа воздуха. Цвет вещества варьируется от пурпурно-красного до фиолетового.
Красный фосфор, в отличие от своего более известного «собрата», белого фосфора, является твердым веществом, не люминесцирует, практически ни в чем не растворим (ни в воде, ни в органических растворителях, ни в сероуглероде). Он не ядовит, самовоспламеняется на воздухе только при температуре +240-260 °С (на самом деле воспламеняется не сам красный фосфор, а его пары, которые после охлаждения превращаются в белый огнеопасный фосфор).
Плотность красного фосфора выше, чем у белого и равна 2,0 – 2,4 г/см3 (в зависимости от конкретной модификации).
На воздухе красный фосфор поглощает влагу, окисляется, превращаясь в оксид; продолжая впитывать влагу, переходит в густую фосфорную кислоту («отмокает»). Ввиду этого, реактив следует герметично укупоривать, лишая доступа к воздушной влаге. При нагревании красный фосфор не плавится, а возгоняется (испаряется). После конденсации пары вещества превращаются в белый фосфор.
Применение красного фосфора
Красный фосфор практически не токсичен и гораздо безопаснее  в работе и хранении, чем белый фосфор. Поэтому в промышленном производстве фосфидов, фосфоросодержащих удобрений, разных производных фосфорной кислоты чаще всего используют красный фосфор.
в работе и хранении, чем белый фосфор. Поэтому в промышленном производстве фосфидов, фосфоросодержащих удобрений, разных производных фосфорной кислоты чаще всего используют красный фосфор.
Сам красный фосфор в основном применяется для изготовления спичек. Он входит в «тёрочную» смесь, которую наносят на коробок. Также его используют в смазочных материалах, в зажигательных составах, топливе, в производстве ламп накаливания.
Не знаете, где купить красный фосфор?
Купить красный фосфор и различные другие химреактивы можно в одном из крупнейших магазинов оборудования для лабораторий, «ПраймКемикалсГрупп». У нас доступные цены и удобная доставка по Москве и области, а квалифицированные менеджеры помогут сделать выбор.
Опубликовано: 02.06.2022
Обновлено: 07.06.2022
Кол-во просмотров: 4070
Читать: 4 минут
Красный фосфор – вещество, которое является модификацией химического элемента фосфора. Другие его аллотропические формы – белый, чёрный, металлический. Данный фосфор неядовитый, неогнеопасный, не светится в темноте. Красное вещество обладает особыми характеристиками, и они подробно рассмотрены в этой статье.
Свойства
Красный фосфор, формула которого Рn, также называется фиолетовым и считается самой стабильной модификацией химического элемента, если рассматривать термодинамические свойства. Разные аллотропические формы обладают различными химическими и физическими характеристиками, что обусловлено разницей в строении. Так, красный фосфор имеет атомную кристаллическую решётку, а белый – молекулярную.
Из-за сложной структуры, состоящей из связанных пирамидально атомов, с другими веществами фосфор вступает в реакции слабо. Этот неорганический естественный полимер может синтезироваться разными способами. И свойства красного фосфора зависят от метода его получения. Их реально прогнозировать, меняя катализаторы, освещение и температуру при процессах производства.
Красный фосфор запаха не имеет. Его цвет зависит от метода производства, а также степени измельчения. Оттенок варьируется от алого до фиолетового. Литое вещество насыщенно-фиолетовое, с медным тоном и блеском металла.
Красный фосфор – какой формы вещество? Он твёрдый, не обладает способностью люминесцировать, то есть создавать свечение в темноте. Токсичным, ядовитым химический элемент в такой модификации не является. Плотность вещества составляет от 2 г/см3 до 2,4, в зависимости от условий получения.
Интересный факт! Всего открыто больше двух десятков модификаций фосфора, но стабильными являются только 4: металлический, чёрный, белый и красный.
Характеристики
Характеристика красного фосфора сильно отличается от описания его ближайшего «родственника» – белого фосфора. Вещество неогнеопасное, воспламеняться может только при термическом воздействии – нагреве до температуры от +240 градусов и выше. Причём загорается не сам химический элемент, а его пары, и после охлаждения они трансформируются в белый фосфор, обладающий высокой огнеопасностью.
Вещество практически ни в чём не растворяется, не вступает во взаимодействие ни с сероуглеродом, ни с водой, ни с органическими растворителями. Лишь в висмуте, свинце и некоторых других расплавленных металлах красный фосфор может растворяться. Такую особенность в химии используют, если необходимо производство красного фосфора в форме крупных кристаллов.

Воспламенение вещества в обычных условиях не происходит, но возможно при ударах или активном трении. Красный фосфор поглощает влагу из атмосферного воздуха. При взаимодействии с кислородом окисляется, становится оксидом. При дальнейшем впитывании H2O вещество отмокает, становясь густой фосфорной кислотой. Из-за таких особенностей красный фосфор поставляется и перевозится только в герметичных упаковках для исключения взаимодействия с воздушной влагой.
При нагревании вещество не расплавляется, а испаряется. Когда пары после конденсации охлаждаются, они превращаются в другую форму фосфора – белую.
Получение
Впервые в 1847 году фосфор был синтезирован химиком из Австрии А. Шрёттером. Это произошло в Швеции, и тогда учёный нагрел до 500℃ находящийся в запаянной ампуле из стекла белый фосфор вместе с угарным газом.
Сегодня получение красного фосфора осуществляется похожим методом. Химики также значительно нагревают белый фосфор, доводя его температуру до 250-300 градусов Цельсия и лишая доступа воздуха. Термическое воздействие должно быть довольно продолжительным – несколько часов. Для производства используется разное оборудование, и в нём также есть запаянные стеклянные ампулы. Иногда для ускорения реакции превращения применяют ионизирующее излучение.

Применение красного фосфора
Красный фосфор используется более широко и часто, чем другие химические соединения, так как он менее активен и ядовит, более стабилен и безопасен в хранении, транспортировке и применении. Основная сфера – промышленность.
Применение красного фосфора осуществляется при:
- Производстве фосфидов. Это бинарные соединения, в которых степень окисления фосфора отрицательная. Фосфиды магния, алюминия и прочих металлов применяются для фумигации – обработки поверхностей против разных насекомых-вредителей, например, моли. Также химические соединения этой категории могут использоваться в солнечных батареях, сцинтилляционных счётчиках.
- Изготовлении фосфоросодержащих удобрений. Они используются для обработки почв с целью улучшения качества грунта и повышения всхожести высаживаемых культур.
- Получении фосфорорганических соединений, например, производных фосфорной кислоты. Такие вещества используются на производствах: текстильных, химических.
- Производстве пиротехники. Фосфор вводят в состав дымозажигательных, дымовых, зажигательных смесей, а также накольных и тёрочных.
- Изготовлении спичек. На коробки наносится специальная тёрочная смесь из красного фосфора.
- Выпуске смазочных материалов, применяемых для уменьшения степени износа деталей из-за трения.
- Военном оснащении. Красный фосфор включается в состав зажигательных снарядов.
- Изготовлении ламп накаливания. Фосфор обеспечивает их свечение.
- Производстве фосфатных связующих. Они входят в составы клеев, используются для холоднотвердеющих смесей, литья и формовки деталей из металлов.
- Нанесении электролюминесцентных покрытий, применяемых для оформления подсветок, систем освещения.
- Реакциях органического синтеза – получении соединений с определёнными заданными свойствами.
- Производстве антипиренов – специализированных материалов, обеспечивающих огнезащиту поверхностей.

Красный фосфор – неядовитое, при обычных условиях нетоксичное вещество. Оно не воспламеняется и почти не вступает во взаимодействие с другими химическими соединениями, поэтому используется в разных сферах.
Фосфор — химический элемент № (15). Он расположен в VА группе третьем периоде Периодической системы.
P15+15)2e)8e)5e
На внешнем слое атома фосфора содержатся пять валентных электронов, до его завершения не хватает трёх электронов. Поэтому в соединениях с металлами и водородом фосфор проявляет степень окисления (–3), а при взаимодействии с более электроотрицательными элементами: кислородом, фтором и другими — положительные степени окисления ( +3) или (+5).
В атоме фосфора больше электронных слоёв по сравнению с атомом азота, поэтому его электроотрицательность, окислительные и неметаллические свойства выражены слабее.
В земной коре фосфор находится в виде фосфатов. Чаще встречается фосфат кальция
Ca3(PO4)2
.
Фосфор — жизненно важный элемент. Он входит в состав нуклеиновых кислот и АТФ, которые необходимы каждой клетке любого живого организма. Фосфат кальция содержится в костной ткани и придаёт ей твёрдость.
Химическому элементу фосфору характерна аллотропия. Он образует несколько простых веществ, отличающихся строением.
Белый фосфор состоит из четырёхатомных молекул
P4
.

Рис. (1). Молекула белого фосфора
Он представляет собой белое (с жёлтым оттенком), похожее на воск вещество, которое светится в темноте из-за окисления кислородом воздуха.
Как все молекулярные соединения, белый фосфор летуч. Он имеет чесночный запах. Не растворяется в воде, но растворяется в сероуглероде. Белый фосфор очень ядовит. В порошкообразном состоянии может самовоспламеняться. Хранят его под водой.
Красный фосфор имеет атомную кристаллическую решётку.

Рис. (2). Строение красного фосфора
Красный фосфор представляет собой порошок и по своим свойствам резко отличается от белого. Он не имеет запаха, не растворяется в воде и в сероуглероде. Неядовит. Активность красного фосфора ниже, чем белого.
Аллотропные модификации фосфора взаимопревращаемы. Белый фосфор превращается в красный на свету или при длительном нагревании без доступа воздуха. Красный фосфор при сильном нагревании и охлаждении паров превращается в белый.
Химические свойства разных аллотропных модификаций фосфора похожи. Белый фосфор более активен и вступает в реакции легче.
Окислительные свойства фосфор проявляет в реакциях с активными металлами:
Полученные соединения называются фосфидами (
Na3P
— фосфид натрия).
В отличие от азота фосфор не соединяется с водородом.
Восстановительные свойства фосфор проявляет в реакции с кислородом. Белый фосфор самовоспламеняется на воздухе, а красный загорается при нагревании. При этом образуется густой белый дым оксида фосфора(V):
Красный фосфор используется при изготовлении спичек.
Источники:
Рис. 1. Молекула белого фосфора © ЯКласс
Рис. 2. Строение красного фосфора © ЯКласс